Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Prenatální období z pohledu onkogenetika Plevová P.

2
1) Prenatálně diagnostikovaná nádorová onemocnění Kongenitální peribronchiální myofibroblastický tumor. Calvo-Garcia et al. Pediatr Radiol (2014) 44:479–483. Epulis http://anesthesia- resident.blogspot.cz/2013 /05/image-of-month.html
 44:479–483. Epulis resident.blogspot.cz/2013 /05/image-of-month.html.")
3
Syndromy predispozice k nádorovým onemocněním s manifestací v dospělém věku Hereditární karcinom prsu a vaječníků (BRCA1, BRCA2, CHEK2) Hereditární nepolypózní kolorektální karcinom (MLH1, MSH2, MSH6, PMS2) Hereditární difúzní karcinom žaludku (CDH1) Hereditární leiomyomatóza/karcinom ledviny (FH) Syndrom familiárního melanomu/ dysplastických névů (CKN2A) Peutz-Jeghersův syndrom (STK11) Hereditární chronická pankreatitis/karcinom pankreatu (CFTR, SPINK1, PRSS1) Cowdenův syndrom (PTEN) Hereditární papilární karcinom ledviny (MET)
 Hereditární nepolypózní kolorektální karcinom (MLH1, MSH2, MSH6, PMS2) Hereditární difúzní karcinom žaludku (CDH1) Hereditární leiomyomatóza/karcinom ledviny (FH) Syndrom familiárního melanomu/ dysplastických névů (CKN2A) Peutz-Jeghersův syndrom (STK11) Hereditární chronická pankreatitis/karcinom pankreatu (CFTR, SPINK1, PRSS1) Cowdenův syndrom (PTEN) Hereditární papilární karcinom ledviny (MET)")
4
s manifestací v dětském i dospělém věku Neurofibromatóza typu 1 a 2 (NF1, NF2) Hippel-Lindauova choroba (VHL) Tuberózní skleróza (TSC1, TSC2, TSC3) Familiární adenomatózní polypóza (APC) von Syndrom Li-Fraumeni (TP53) Syndromy mnohočetné endokrinní neoplázie typ 1, 2 (MEN1, RET) Gorlinův syndrom (PTCH) Hereditární paragangliom/feochromocytom (SDHA, SDHB, SDHC, SDHD, SDHA2F) Ataxia telangiextasia (ATM) Bloomův syndrom (BLM) Fanconiho anémie (FANCA-FANCN) Xeroderma pigmentosum (XPA, ERCC3, XPC, ERCC2, XPE, ERCC4, ERCC5, POLH) Wernerův syndrom (WRN) Syndrom Proteus (AKT1) Carneyho syndrom (PRKAR1A)
 Hippel-Lindauova choroba (VHL) Tuberózní skleróza (TSC1, TSC2, TSC3) Familiární adenomatózní polypóza (APC) von Syndrom Li-Fraumeni (TP53) Syndromy mnohočetné endokrinní neoplázie typ 1, 2 (MEN1, RET) Gorlinův syndrom (PTCH) Hereditární paragangliom/feochromocytom (SDHA, SDHB, SDHC, SDHD, SDHA2F) Ataxia telangiextasia (ATM) Bloomův syndrom (BLM) Fanconiho anémie (FANCA-FANCN) Xeroderma pigmentosum (XPA, ERCC3, XPC, ERCC2, XPE, ERCC4, ERCC5, POLH) Wernerův syndrom (WRN) Syndrom Proteus (AKT1) Carneyho syndrom (PRKAR1A)")
5
s manifestací v dětském věku Syndrom WAGR (Wilms-aninridia-genitourinary malformation-mental retardation) (WT1, PAX6) Beckwith-Wiedemannův syndrom (CDKN1C, H19, KCNQ1, NSD1) Idiopatická hemihypertrofie a aniridie (WT1) Denys-Drashův syndrom (WT1) Syndrom Simpson-Golabi-Behmel (GPC3) Perlmanův sy (GPC3) Familiární retinoblastom (RB1) Ataxia telangiextasia (ATM) Bloomův syndrom (BLM) Nijmegen Breakage syndrom (NBS1) Seckelův syndrom (ATR) Rothmund-Thomsonův syndrom (RECQL4) Syndrom Costello (HRAS) Hereditární neuroblastom (PHOX2B, ALK) Syndrom predispozice k rhabdoidním tumorům (SMARCB1) Syndrom konstituční insuficience systému oprav chybného párování bází (PMS2, MSH6, MLH1, MSH2)
 (WT1, PAX6) Beckwith-Wiedemannův syndrom (CDKN1C, H19, KCNQ1, NSD1) Idiopatická hemihypertrofie a aniridie (WT1) Denys-Drashův syndrom (WT1) Syndrom Simpson-Golabi-Behmel (GPC3) Perlmanův sy (GPC3) Familiární retinoblastom (RB1) Ataxia telangiextasia (ATM) Bloomův syndrom (BLM) Nijmegen Breakage syndrom (NBS1) Seckelův syndrom (ATR) Rothmund-Thomsonův syndrom (RECQL4) Syndrom Costello (HRAS) Hereditární neuroblastom (PHOX2B, ALK) Syndrom predispozice k rhabdoidním tumorům (SMARCB1) Syndrom konstituční insuficience systému oprav chybného párování bází (PMS2, MSH6, MLH1, MSH2)")
6
Tuberózní skleróza 50-85% kardiálních rabdomyomů souvisí s TS (kazuisticky popsán RM u Beckwith Wiedemannova sy) AD dědičnost Mutace genů TSC1 (pro hamartin), TSC2 (pro tuberin) Testování : OLG FN Olomouc aktivace signální dráhy mTOR (mammalian target of rapamycin) s rozvojem hamartomatózních lézí http://www.novusbio.com/mTOR-pathway Postnatální projevy Angiolipom ledviny Adenoma sebaceum Hypopigmentované skvrny Šangrénové skvrny Unguální keratom Kortikální tubery Subependy- mální nodul Kardiální rhabdomyom
 AD dědičnost Mutace genů TSC1 (pro hamartin), TSC2 (pro tuberin) Testování : OLG FN Olomouc aktivace signální dráhy mTOR (mammalian target of rapamycin) s rozvojem hamartomatózních lézí Postnatální projevy Angiolipom ledviny Adenoma sebaceum Hypopigmentované skvrny Šangrénové skvrny Unguální keratom Kortikální tubery Subependy- mální nodul Kardiální rhabdomyom")
7
Kardiální rhabdomyomy – léčba everolimem Věk 1 rok 5 let Po 13 měs. léčby everolimem Tiberio et al. Pediatrics 2011;127:e1335 -e1337 Kazuistiky – regrese srdečního RM u 6-ti letého dítěte (Tiberio et al. Pediatrics 2011) – inoperabilní symptomatický RM (Dogan et al. J Trop Pediatr 2014) – předčasně narozený novorozenec s mnohočetnými RM srdce (a atrézií plicnice a velkým komorovým defektem) (Mlczoch et al. Ultrasound Obstet Gynecol 2014) Většina RM spontánně regreduje během 1. roku života Léčba inhibitory mTOR postnatálně – everolimus (původně léčba subependymálních giant-cell astrocytomů a renálních angiomyolipomů) http://www.selleckchem.com/blog/Everolimus-as- a-mTOR-inhibitor.html
 – inoperabilní symptomatický RM (Dogan et al. J Trop Pediatr 2014) – předčasně narozený novorozenec s mnohočetnými RM srdce (a atrézií plicnice a velkým komorovým defektem) (Mlczoch et al. Ultrasound Obstet Gynecol 2014) Většina RM spontánně regreduje během 1. roku života Léčba inhibitory mTOR postnatálně – everolimus (původně léčba subependymálních giant-cell astrocytomů a renálních angiomyolipomů) a-mTOR-inhibitor.html.")
8
Neurofibromatóza 1. typu Kazuistika McEwing et al. Prenatal Diagnosis 2006;26:1110-1114. 27.t.těh. polyhydramnion, hydrocephalus, kardiomegalie, hypertrofie myokardu 29.t.těh + pleurální efúze bilat. 31.t.těh. + makroglosie, farynx- hypoechogenní masa, ztluštění krku, perikardiální efúze, ascites 32.t.těh. – těhotenství ukončeno Otec: NF1. typu AD dědičnost Gen NF1 (pro neurofibromin) Testování : ÚBLG FN Motol, OLG FN Brno
 Testování : ÚBLG FN Motol, OLG FN Brno.")
9
Neurofibromatóza 1. typu Postnatální projevy Neurofibromy Gliom optického nervu Skvrny „café au lait“ Plexiformní neurinom McEwing et al. Prenatal Diagnosis 2006;26:1110-1114.

10
Neurofibromatóza 1. typu Obstruktivní léze aqueduktu – pilocytární nebo low-grade astrocytomy, gliomy, hamartomy, glióza, gangliomy Marcorelles et al. Clin Neuropathol 2005 Fetální glioneurální hamartom http://sonoworld.com/TheFetus/

11
Hereditární retinoblastom Zárodečné mutace v genu RB1 http://wiki.ggc.edu/wiki/File:Retinoblastoma_beggining_pic.jpg AD, 1:20 000 Testování: OLG FN Brno 40 % dědičných „doubling time“ 7 dní někdy nádory pokročilé již v době porodu časná detekce prenatálně ? prenatální detekce hyperechogenních lézí záleží na velikosti (výška nad 1-2 mm) a morfologii (minimálně elevované versus elevované) Paquette et al. AJP Rep. 2012 Nov;2(1):55-62.
 a morfologii (minimálně elevované versus elevované) Paquette et al. AJP Rep Nov;2(1):")
12
Hereditární retinoblastom – prenatální skríning 6 plodů s rizikem retinoblastomu HR-UZV očí fétu od 16.-18.t.těh. á 4 týdny do 32.t.těh., pak á 2 týdny fetální NMR od 16.-18.t.těh. á 8 týdnů postnatálně potvrzen u 1 novorozence (3 léze, 1 dg. UZV prenatálně (2-3 mm, 37.t.těh.), další 2 nikoli) prenatální UZV a fetální NMR – málo senzitivní pro minimálně elevované léze, UZV senzitivnější pro elevované léze než NMR Paquette et al. AJP Rep. 2012 Nov;2(1):55-62.
, další 2 nikoli) prenatální UZV a fetální NMR – málo senzitivní pro minimálně elevované léze, UZV senzitivnější pro elevované léze než NMR Paquette et al. AJP Rep Nov;2(1):")
13
Hereditární neuroblastom 1-2% nově dg. případů je familiárních – IR 9,7 pro sourozence probanda – časnější věk, mnohočetné primární nádory gen PHOX2B – menšina hereditárních NB gen ALK (anaplastic lymphoma kinase gene) Testování: Gennet Praha http://indianpediatrics.net/mar2002/mar- 308.htm 8 rodin s familiárním neuroblastomem – missense mutace v genu ALK Mosse et al. Nature. Oct 16, 2008; 455(7215): 930–935.
 Testování: Gennet Praha htm 8 rodin s familiárním neuroblastomem – missense mutace v genu ALK Mosse et al. Nature. Oct 16, 2008; 455(7215): 930–935..")
14
Syndrom konstituční insuficience systému oprav chybného párování bází Wimmer et al. J Med Genet 2014;51:355-365 Geny: PMS2: 60% MSH6: 20% MLH1/MSH2: 20% Bialelická mutace genů pro Lynchův syndrom Geny: MLH1/MSH2: 80% MSH6: 15% PMS2: 5% Lynchův syndrom (hereditární nepolypózní kolorektální karcinom) ca coli (27) ca coli (40) ca coli (44) ca endometria (45) ca endometria (50) ca coli (55) ca coli (60) ca coli (53 ca žaludku 55)) ca endometria (43) Testování: PMS2: v ČR neodstupné MLH1, MSH2, MSH6: ÚBLG FN Motol, MOÚ Brno, Agel Nový Jičín
 ca coli (27) ca coli (40) ca coli (44) ca endometria (45) ca endometria (50) ca coli (55) ca coli (60) ca coli (53 ca žaludku 55)) ca endometria (43) Testování: PMS2: v ČR neodstupné MLH1, MSH2, MSH6: ÚBLG FN Motol, MOÚ Brno, Agel Nový Jičín.")
15
Baas et al. Eur J Hum Genet. Jan 2013; 21(1): 55–61. Syndrom konstituční insuficience systému oprav chybného párování bází

16
Vrozené vývojové vady: Ageneze corporis callosi s/bez heterotopie šedé hmoty mozkové Kavernózní hemangiomy mozku Kapilární hemangiom kůže Asplenie Defekt komorového septa 2013: 146 případů Nádory: Zhoubné: Hematologické: HNL, ALL, AML (CML) Tumory mozku: high grade gliomy, astrocytomy, CNS- PNET, meduloblastom Karcinomy: tlusté a tenké střevo, endometrium, uropoe- tický systém, vaječníky Jiné: neuroblastom, Wilmsův tumor, rabdomyosarkom, osteosarkom Nezhoubné: Polypy v colon, jinde v GIT Pilomatrikomy Syndrom konstituční insuficience systému oprav chybného párování bází Wimmer et al. J Med Genet 2014;51:355-365

17
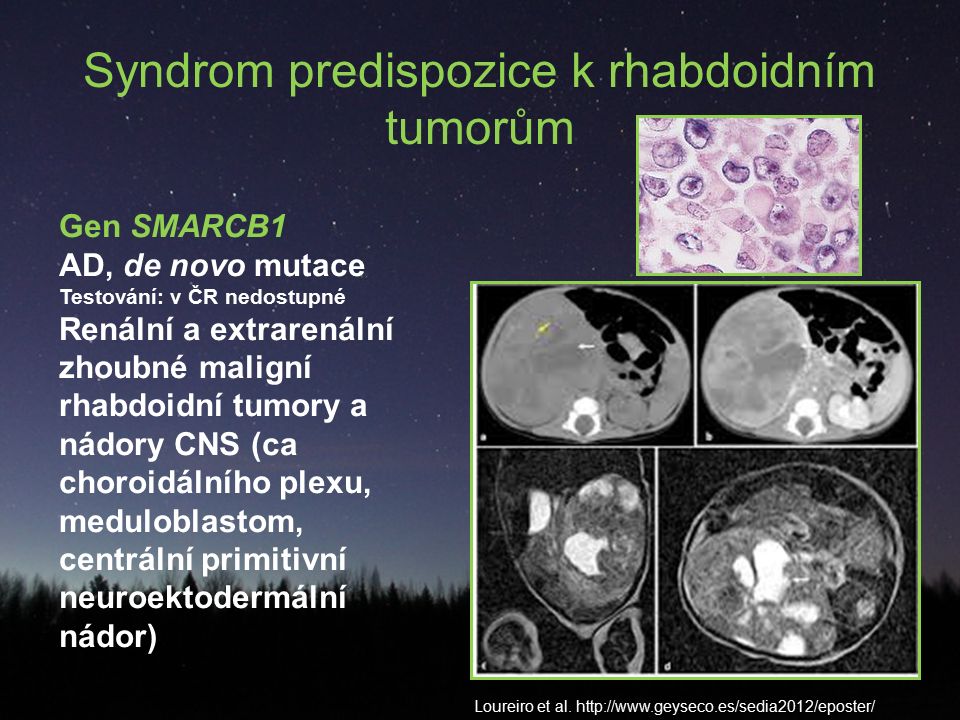
Syndrom predispozice k rhabdoidním tumorům Gen SMARCB1 AD, de novo mutace Testování: v ČR nedostupné Renální a extrarenální zhoubné maligní rhabdoidní tumory a nádory CNS (ca choroidálního plexu, meduloblastom, centrální primitivní neuroektodermální nádor) Loureiro et al. http://www.geyseco.es/sedia2012/eposter/
18
Familiární nefroblastom Prenatálně dg. bilaterální nefroblastomatóza Otec a jeho 4 sourozenci nefroblastomatóza nebo Wilmsův tumor ledviny Array-CGH – duplikace v oblasti 2p24.3 o velikosti 569 kb zahrnující geny DDX1 a MYCN Fievet et al. Eur J Med Genet 2013 Dec;56(12):643-7. http://www.ultrasoundcases.info/ Nefroblastom Nefroblastomatóza – difúzní nebo multifokální persistence nefrogenních zbytků v ledvinách (tj. ložisek metanefrogenního blastému) po 36. t. těh. (tj. po dokončení nefrogeneze) – riziko maligní transformace do Wilmsova tumoru (předchází 41 % unilat. WT a 90 % bilat. WT) (Anand et al. In J Med Paed Oncol 2012;33:242-9.)
: Nefroblastom Nefroblastomatóza – difúzní nebo multifokální persistence nefrogenních zbytků v ledvinách (tj. ložisek metanefrogenního blastému) po 36. t. těh. (tj. po dokončení nefrogeneze) – riziko maligní transformace do Wilmsova tumoru (předchází 41 % unilat. WT a 90 % bilat. WT) (Anand et al. In J Med Paed Oncol 2012;33:242-9.).")
19
2) VVV a riziko nádorových onemocnění Souvislost mezi narušením normálních vývojových procesů a onkogenezí Děti s vrozenou vývojovou vadou mají 3 x vyšší riziko rozvoje nádorů (nádory z germinálních bb., retinoblastom, sarkomy měkkých tkání, leukémie, neuroblastom) Downův syndrom – leukémie (nižší riziko jiných nádorů) Hemihypertrofie, Beckwith-Weidemanův syndrom - Wilmsův tumor Aniridie - Wilmsův tumor, tumory CNS Kloaka, hypoplázie končetin - teratomy
 VVV a riziko nádorových onemocnění Souvislost mezi narušením normálních vývojových procesů a onkogenezí Děti s vrozenou vývojovou vadou mají 3 x vyšší riziko rozvoje nádorů (nádory z germinálních bb., retinoblastom, sarkomy měkkých tkání, leukémie, neuroblastom) Downův syndrom – leukémie (nižší riziko jiných nádorů) Hemihypertrofie, Beckwith-Weidemanův syndrom - Wilmsův tumor Aniridie - Wilmsův tumor, tumory CNS Kloaka, hypoplázie končetin - teratomy")
20
Birth Defects Group No. of Cases With Birth Defects a a IRR95% CI Central nervous system173.612.10, 5.79 Neural tube 43.030.83, 7.78 Eye and ear63.471.27, 7.56 Anophthalmia/micropht halmia 56.912.24, 16.14 Cardiac and circulatory913.502.81, 4.31 Conotruncal 73.141.26, 6.47 Septal 603.052.32, 3.94 Left ventricular outflow tract 544.223.16, 5.53 Respiratory53.581.16, 8.36 Oral clefts112.691.34, 4.82 Gastrointestinal131.690.90, 2.89 Gastrointestinal atresia/stenosis 52.560.83, 5.97 Genitourinary342.371.64, 3.32 Musculoskeletal50.880.29, 2.06 Limb reduction defects10.800.02, 4.49 Abdominal wall defects32.260.47, 6.62 Chromosomal b b 5515.5211.66, 20.27 Any monitored defect2342.862.49, 3.28 Carozza et al. Are Children With Birth Defects at Higher Risk of Childhood Cancers? Am. J. Epidemiol. (2012) 175 (12): 1217-1224. Riziko nádoru v dětském věku podle charakteru VVV
 175 (12): Riziko nádoru v dětském věku podle charakteru VVV.")
21
3) Prenatální období jako kritické pro rozvoj nádorů postnatálně Kouření otce v období kolem koncepce – 1,5-2x zvýšené riziko, epigenetické změny Kouření marihuany matkou v 1. trimestru těh. – 5x zvýšené riziko neuroblastomu Milne et al. J Epidemiol 2012 Jan 1;175(1):43-53. Bluhm et al. Cancer Causes Control. 2006 Jun;17(5):663-9. Neepiteliální ovariální nádory - 2x vyšší riziko u žen narozených předčasně (pod 37.t.těh.), pro stromální nádory vaječníků riziko 4x vyšší (pro nádory ze zárodečných buněk nezvýšené ) Sieh et al. Gynecol Oncol. 2014 May;133(2):293-7.
: Bluhm et al. Cancer Causes Control Jun;17(5): Neepiteliální ovariální nádory - 2x vyšší riziko u žen narozených předčasně (pod 37.t.těh.), pro stromální nádory vaječníků riziko 4x vyšší (pro nádory ze zárodečných buněk nezvýšené ) Sieh et al. Gynecol Oncol May;133(2):")
22
Závěr Geneticky podmíněná nádorová onemocnění jsou vzácná, lze očekávat manifestaci syndromů, které se prozatím prenatálně nemanifestovaly Souvislost mezi narušením normálních vývojových procesů a onkogenezí Prenatální období je rizikové z hlediska rozvoje nádorů postnatálně

23
Děkuji Vám za pozornost

Podobné prezentace



















