Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Cystická fibróza (Cystic fibrosis transmembrane regulator)
Veronika Lišková MBB, 2. ročník Univerzita Palackého v Olomouci

2
Co je cystická fibróza? Dědičné onemocnění
Vyskytuje se přibližně u jednoho z 2000 – 2500 živě narozených dětí bílého plemene V ČR – dětí ročně (nyní cca 500) Polovina postižených se dožívá 32 let
 Polovina postižených se dožívá 32 let.")
3
postihuje a likviduje funkce plic, pankreatu, jater i střev
Nejčastější smrtelnou geneticky podmíněnou chorobou indoevropského obyvatelstva

4
Příznaky a projevy CF Tvorba hustého sekretu → parazité → záněty
Aspergilus fumigatus ( Burkholderia cepacia (

5
Graf srovnávající výskyt jednotlivých patogenů v plicích pacientů s cystickou fibrózou

6
Dýchací soustava ucpává dýchací trubice a snižuje kapacitu dýchacího stromu

7
Trávicí systém ucpané vývody slinivky břišní a žlučníku
Malabsorbce, cirhóza jater Potíže se vstřebáváním vitamínů D, E a K

8
Reprodukční soustava Muži – vrozená absence spermatických vývodů varlat (98 %) Ženy – snížená plodnost
 Ženy – snížená plodnost.")
9
Slaný pot Nosní polypóza
Potní žlázy neprodukují hlen → produkce ↓ množství potu, ALE s ↑ obsahem Na, Cl Ztráta soli → negativní vliv na metabolismus vody Nosní polypóza Starší a dospívající děti Nutná operace

10
Endokrinní soustava a růst
Slinivka břišní – Langerhansovy ostrůvky → diabetes Absence vit. D → osteoporóza Zduření prstů Malabsorbce → nedostaktek živin → podvýživa, chudý růst

11
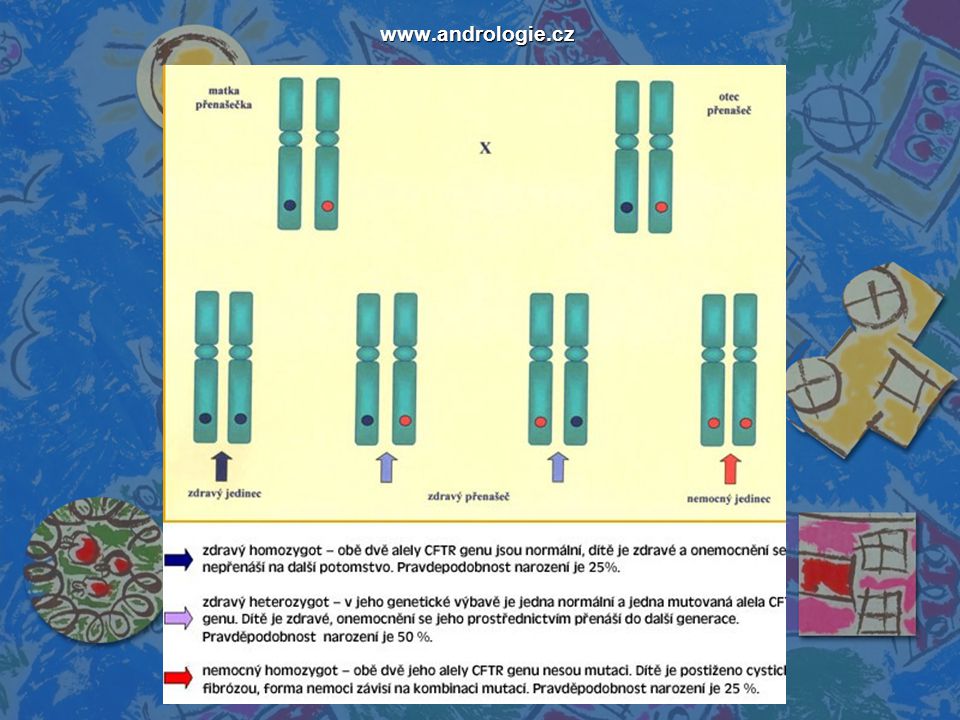
Pohled genetiky Dědičné onemocnění Na autozomu
Recesivní genetická mutace jediného genu → projev u recesivního homozygota → rodiče heterozygoté (každý pár) → 25% pravděpodobnost → projev v šířce rodokmenu (čeká na šanci, kdy se vyštěpit)
 → 25% pravděpodobnost → projev v šířce rodokmenu (čeká na šanci, kdy se vyštěpit)")
13
Molekulárně metabolický charakter
U 1 z 2000 – 2500 narozených dětí bílého plemene Indoevropské národy – 3-4 % heterozygoté Asijské a africké národy – 0,001 % heterozygotů !!!Nejčastější genová mutace na světě!!!

14
Molekulární podstata Buňky epitelu jsou defektní v transmembránovém přenosu iontů Porucha membránového kanálu pro Clˉ ionty Do buňky prochází více Na s vodou Hlen přestává být hypotonický → nedochází k aktivaci antimikrobiálních peptidů

15
Ztráta enormního množství Cl pocením u postižených dětí → „slané děti“
Ztráta enormního množství Cl pocením u postižených dětí → „slané děti“

16
Na 7. autozomu – dlouhé ramínko q v proužku 7q31
Gen pro cystickou fibrózu – CFTR gene (cystic fibrosis transmembrane regulator gene) Na 7. autozomu – dlouhé ramínko q v proužku 7q31
 Na 7. autozomu – dlouhé ramínko q v proužku 7q31.")
17
CFTR gene z více jak 250 000 nukleotidů 26 exonů
Informace pro syntézu proteinu – membránový chloridový kanál Z AK

18
CFTR gene Přes 1350 mutací – 5 tříd
V ČR - ∆F508 (71,6 %) → ztráta 3 neukkleotidů → ztráta AK fenylalanin → nemůže vázat cAMP mutace → neopustí ER → nedojde k transportu k buněčné membráně
 → ztráta 3 neukkleotidů → ztráta AK fenylalanin → nemůže vázat cAMP. mutace → neopustí ER → nedojde k transportu k buněčné membráně.")
19
CFTR protein Karikaturní vykazování molekulární struktury CFTR bílkoviny (

20
CFTR protein

21
Vznik delece pravděpodobně před 52 000 lety
Různé mutace genu ovlivňují fyziologickou funkci tohoto proteinu různou měrou a projevují se rozdílnou expresivitou Vznik delece pravděpodobně před lety Populace podobná dnešním Baskům

22
Diagnostická kritéria CF
Diagnostika Diagnostická kritéria CF klinické příznaky a/nebo pozitivní rodinná anamnéza pozitivní novorozenecký screening + opakovaně pozitivní potní test průkaz mutací obou alel CFTR genu pozitivní výsledek transepiteliálního rozdílu potenciálů

23
Den s CF

24
Zdroje Jan Šmarda: Člověk v proudu dědičnosti

Podobné prezentace

















 Zuzana Hofová.>")

. Prognózování GPS a genetické poradenství Principem genetického prognózování je předpovědění vzniku určitého.>")