Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Buněčný stres a adaptace
Mgr. Gabriela Rylová Laboratoř experimentální medicíny

2
Úvod: buněčná odpověď na stres
Normální buňka má danou funkci a strukturu, jenž je podmíněná genetickým programem jejího metabolismu, diferenciace a specializace. HOMEOSTÁZE: vyvážený stav buňky Fyziologický stres a patologické podněty adaptace pozměněný stav buňky zajišťující ochranu viability buněk, modulace jejích funkcí jako odpověď na některé podněty Mechanismy adaptace: - hyperplazie, hypertrofie, atrofie, metaplazie - zvýšená syntéza proteinů(chaperony, kolagen), lipidů, karbohydrátů - kalcifikace – ukládání vápníku
, lipidů, karbohydrátů. - kalcifikace – ukládání vápníku.")
3
Adaptace růstu a diferenciace
Buňka odpovídá na zvýšené požadavky hypertrofií nebo hyperplazií, na nedostatek živin atrofií, metaplazie - přechod z jednoho typu buněk na jiný mnoho molekulárních mechanismů: indukce faktory samotné buňky nebo okolních buněk zvýšení proliferace buňek (endometrium) nadprodukce určitých proteinů (kolagen u fibrózy) zvýšení proteinové syntézy (myocyty)
 nadprodukce určitých proteinů (kolagen u fibrózy) zvýšení proteinové syntézy (myocyty)")
4
Hyperplazie Hyperplazie = zvýšení počtu buněk v orgánu nebo tkáni
často se vyskytuje spolu s hypertrofií, spuštěné stejným mechanismem fyziologická hyperplazie hormonální hyperplazie: zvyšuje funkční kapacitu tkáně (proliferace žlázového epitelu prsou v pubertě a těhotenství) kompenzační hyperplazie: obnovuje tkáň po poškození nebo částečné resekci (játra, ledviny, obnova tkáně při zranění) patologická hyperplazie nadměrná hormonální stimulace RF (hyperplazie endometria vlivem estrogenů, prostatická hyperplazie vlivem androgenů) Mechanismus hyperplazie: zvýšená produkce růstových faktorů, zvýšení počtu receptorů pro RF, aktivace signálních drah produkce transkripčních faktorů zapínání genů pro RF, receptory RF, regulátory buněčného cyklu proliferace
 kompenzační hyperplazie: obnovuje tkáň po poškození nebo částečné resekci (játra, ledviny, obnova tkáně při zranění) patologická hyperplazie. nadměrná hormonální stimulace RF (hyperplazie endometria vlivem estrogenů, prostatická hyperplazie vlivem androgenů) Mechanismus hyperplazie: zvýšená produkce růstových faktorů, zvýšení počtu receptorů pro RF, aktivace signálních drah produkce transkripčních faktorů. zapínání genů pro RF, receptory RF, regulátory buněčného cyklu. proliferace.")
5
Hyperplazie vs. neoplazie
Hyperplazie – fyziologická proliferace, ukončením působení RF nebo hormonů se buňky navracejí do normálního stavu Neoplazie – abnormální proliferace, ztráta kontroly proliferace, ztráta zpětnovazebné smyčky, aktivace pozitivních mitotických signálů Hyperplastické buňky jsou náchylné k neoplazii

6
Hypertrofie Hypertrofie = zvětšení velikosti buněk, buňky se nedělí, jen zvětšují svůj objem na základě syntézy více strukturálních komponent Fyziologická hypertrofie (hypertrofie dělohy a prsou při těhotenství, posilování) Patologická hypertrofie (zvýšení počtu myofilament a proteinů v buňce při zátěži-hypertenze, vadné chlopně) Mechanismus hypertrofie: U srdce: signální dráhy indukce genů syntéza proteinů TF, RF, vazoaktivní agens, produkce kontraktilních proteinů
 Patologická hypertrofie (zvýšení počtu myofilament a proteinů v buňce při zátěži-hypertenze, vadné chlopně) Mechanismus hypertrofie: U srdce: signální dráhy indukce genů syntéza proteinů. TF, RF, vazoaktivní agens, produkce kontraktilních proteinů.")
7
Spouštěcí mechanismy hypertrofie:
Mechnické spouštěče ( stah) Trofické spouštěče (polypeptidové RF, vazoaktivní agens – přenos signálu do jádra genová exprese Pokud srdce není schopno dlouhodobě kompenzovat zátěž dojde k degenerativním změnám v myokardiálních vláknech –lyze, ztráta kontraktilních elementů apoptóza, nekróza
 Trofické spouštěče (polypeptidové RF, vazoaktivní agens – přenos signálu do jádra genová exprese. Pokud srdce není schopno dlouhodobě kompenzovat zátěž dojde k degenerativním změnám v myokardiálních vláknech –lyze, ztráta kontraktilních elementů apoptóza, nekróza.")
8
Atrofie Atrofie=zmenšování buněk, redukce buněčných komponent
- př. atrofická svalová buňka úbytek mitochondrií, endoplasmatického retikula, nastavení nové rovnováhy - má omezenou funkci, ale není mrtvá - zvýšení katabolismu Fyziologická (součástí embryogeneze, zmenšení dělohy po porodu) Patologická – podle příčin (snížená zátěž, ztráta inervace, ischemie, nedostatečná výživa, ztráta endokrinní stimulace, stárnutí, tlak)
 Patologická – podle příčin (snížená zátěž, ztráta inervace, ischemie, nedostatečná výživa, ztráta endokrinní stimulace, stárnutí, tlak)")
9
Mechanismus atrofie: klíčová role zvýšené degradace proteinů
lysozomy (kathepsiny) ubikvitin – proteazomová degradace (řízená značením proteinů ubikvitinem s následnou degradací v proteazomu) autofagie ( autofagní vakuoly navázané na membráně, obsahují fragmenty buněčných komponent-štěpení)
 ubikvitin – proteazomová degradace (řízená značením proteinů ubikvitinem s následnou degradací v proteazomu) autofagie ( autofagní vakuoly navázané na membráně, obsahují fragmenty buněčných komponent-štěpení)")
10
Metaplazie Metaplazie=reverzibilní změna při které je jeden typ adultní buňky nahrazen jiným typem, odolnějším vůči stresu drsný skvamózní epitel nahrazuje fragilní sloucový epitel jako odpověď na podráždění (př. epitel kuřáků –řasinkový sloupcový epitel vrstevnatý skvamózní epitel; nedostatek vitaminu A;mesenchymální metaplasie, výskyt kostní a chrupavčité tkáně ve svalové tkáni po zlomenině) Skvamózní buňky jsou sice odolnější, ale ztrácí schopnost mukózní sekrece Přetrvávající vlivy vedoucí k metaplazii mohou indukovat maligní transformaci
 Skvamózní buňky jsou sice odolnější, ale ztrácí schopnost mukózní sekrece. Přetrvávající vlivy vedoucí k metaplazii mohou indukovat maligní transformaci.")
11
Příčiny buněčného poškození
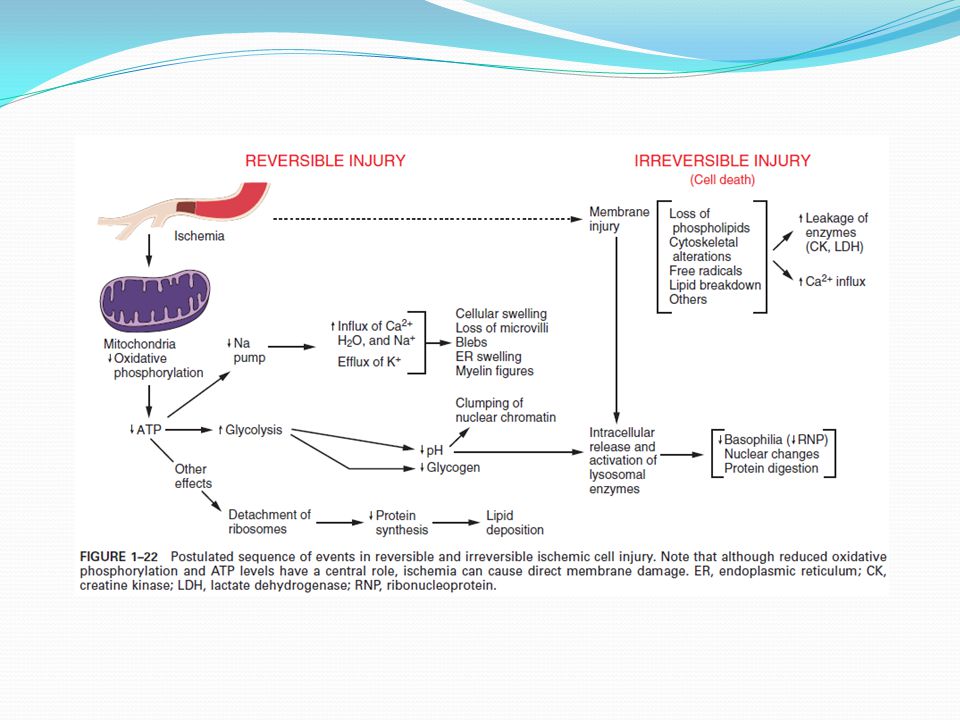
Reversibilní poškození-funkční a morfologické změny, vratné po odeznění poškozujícího podnětu Nevratné poškození buněčná smrt - ?prahový bod? - některé strukturní (poškozené MTCH) a funkční (ztráta permeability membrány) změny indikují ireversibilní poškození
 a funkční (ztráta permeability membrány) změny indikují ireversibilní poškození.")
12
Příčiny buněčného poškození
Hypoxie= nedostatek kyslíku, který způsobuje buněčné poškození snižující aerobní oxidativní respiraci x ischemie= nedostatečné zásobení krví –kyslík, metabolické substráty, glukóza; vážnější poškození selhání srdce a plic, anemie, otrava CO V závislosti na typu hypoxie adaptace, poškození nebo smrt Fyzikální příčiny=mechanické poškození, extrémní teploty,změny tlaku, záření, elektrický šok Působení chemických látek= některé jednoduché látky jako glukóza, soli nebo kyslík můžou být ve vyšších koncentracích toxické -jedy: arsen, kyanid, rtuť,znečištění, insekticidy,herbicidy, CO, azbest,alkohol, drogy, léčiva

13
Příčiny buněčného poškození
Infekce=viry, baterie, fungi, paraziti Imunitní reakce=anafylaxe, autoimunitní choroby Genetické změny=od chromozomálních abnormalit (Downův sy) po jednoaminokyselinové změny (anémie), enzymatické abnormaity-nedostatečná funkce, ztráta funkce nevyvážená výživa= nedostatek vitamínů x nadměrné ukládání tuků, obesita, diabetes, ateroskleróza
 po jednoaminokyselinové změny (anémie), enzymatické abnormaity-nedostatečná funkce, ztráta funkce. nevyvážená výživa= nedostatek vitamínů x nadměrné ukládání tuků, obesita, diabetes, ateroskleróza.")
14
Mechanismus buněčného poškození
Odpověď na poškození závisí: na typu, intenzitě a trvání stresu na typu, stavu a adaptabilitě buňky Poškození Aerobní respirace, MTCH oxidativní fosforylace a produkce ATP Integrita membrán Syntéza proteinů Cytoskelet Integrita genetické informace

15
Vyčerpání ATP deplece ATP a snížení syntézy ATP často v důsledku hypoxie a chemického poškození ATP důležité pro množství syntetických a degradačních procesů v buňce ATP produkováno dvěma cestami: Oxidativní fosforylace ADP Glykolýza (glukóza z krevního zásobení nebo rozkladem glykogenu) Snížení zásob ATP na 5-10% má vliv na: Aktivitu ATP poháněných sodíkových pump Na2+ se hromadí v buňce K+ difunduje ven z buňky Příjem vody bobtnání , rozpínání ER Změny energetického metabolismu Nedostatek kyslíku zastavení oxidativní fosforylace přepnutí na anaerobní metabolismus-glykolýza ze zásob glykogenu Metabolity glykolýzy (laktát, fosfáty) snížení pH redukce aktivity mnoha buněčných enzymů
 Snížení zásob ATP na 5-10% má vliv na: Aktivitu ATP poháněných sodíkových pump. - Na2+ se hromadí v buňce. K+ difunduje ven z buňky. Příjem vody bobtnání , rozpínání ER. Změny energetického metabolismu. Nedostatek kyslíku zastavení oxidativní fosforylace přepnutí na anaerobní metabolismus-glykolýza ze zásob glykogenu. Metabolity glykolýzy (laktát, fosfáty) snížení pH redukce aktivity mnoha buněčných enzymů.")
16
Poškození Ca2+ pump influx Ca2+ Poškození aparátu pro syntézu proteinů
oddělení ribosomů od ER, rozdělění polyzomů na monozomy Špatné skládání proteinů

17
Mitochondriální poškození
Jeden z hlavních cílů mnoha typů buněčného poškození (toxiny, hypoxie) Morfologické změny Poškození: zvýšením cytosolického Ca2+ oxidativní stres selhání lipidového metabolismu vznik neselektivních pórů ( kanálky) ve vnitřní membráně MTCH narušení membránového potenciálu smrt buňky uvolnění cytochromu C do cytoplasmy apoptóza
 Morfologické změny. Poškození: zvýšením cytosolického Ca2+ oxidativní stres. selhání lipidového metabolismu. vznik neselektivních pórů ( kanálky) ve vnitřní membráně MTCH narušení. membránového potenciálu smrt buňky. uvolnění cytochromu C do cytoplasmy apoptóza.")
18
Influx intracelulárního Ca2+ a ztráta Ca2+ homeostáze
Nízká koncentrace v buňce, vysoká extracelulární koncentrace MTCH, ER Ischemie, toxiny zvýšení cytosolického Ca2+ (PM, MTCH, ER) aktivace některých enzymů se školdlivými účinky (ATPázy, fosfolipázy,proteázy)+ zvýšená permeabilita MTCH membrány apoptóza Ztráta Ca2+ homeostáze může být reverzibilní i ireverzibilní
 aktivace některých enzymů se školdlivými účinky (ATPázy, fosfolipázy,proteázy)+ zvýšená permeabilita MTCH membrány apoptóza. Ztráta Ca2+ homeostáze může být reverzibilní. i ireverzibilní.")
19
Oxidativní stres Můžou být indukovány mnoha způsoby:
Jako vedlejší produkty buněčného dýchání vznikají volné kyslíkové radikály (reactive oxygen species) Vysoce reaktivní sloučeniny s nepárovým elektronem – reakce s proteiny, lipidy, nukleovými kyselinami Buňka má obranné mechanismy, pokud dojde k nerovnováze, vzniká oxidativní stres Vznikají po působení chem.látek, záření, při stárnutí, ničení mikrobů fagocyty Můžou být indukovány mnoha způsoby: adsorbce záření (UV, rentgenové) metabolizované chemikálie, drogy (CCl4 na CCl3) oxidačně redukční reakce normálního metabolismu ( vznik O2-,H2O2, OH- jako vedlejších produktů buněčného dýchání, O2- u leukocytů při zánětu) Kovy (Fe, Hg) poskytují nebo přijímají volné e-, Fentonova reakce: H2O2+ Fe Fe3+ + OH + OH- NO tvořen endoteliálními buňkami, makrofágy, neurony volný radikál+ vzniká z něj peroxynitrit a další reaktivní formy
 Vysoce reaktivní sloučeniny s nepárovým elektronem – reakce s proteiny, lipidy, nukleovými kyselinami. Buňka má obranné mechanismy, pokud dojde k nerovnováze, vzniká oxidativní stres. Vznikají po působení chem.látek, záření, při stárnutí, ničení mikrobů fagocyty. Můžou být indukovány mnoha způsoby: adsorbce záření (UV, rentgenové) metabolizované chemikálie, drogy (CCl4 na CCl3) oxidačně redukční reakce normálního metabolismu ( vznik O2-,H2O2, OH- jako vedlejších produktů buněčného dýchání, O2- u leukocytů při zánětu) Kovy (Fe, Hg) poskytují nebo přijímají volné e-, Fentonova reakce: H2O2+ Fe2+ Fe3+ + OH + OH- NO tvořen endoteliálními buňkami, makrofágy, neurony. volný radikál+ vzniká z něj peroxynitrit a další reaktivní formy.")
20
Důsledky oxidačního stresu
Peroxidace lipidů v membránách - dvojná vazba nenasycených mastných kyselin je atakována volnými radikály, reakcí vznikají peroxidy (nestabilní, reaktivní, autokatalycké) poškození membrán a organel ukončení vychytávačem volných radikálů (vitamin E) Oxidativní modifikace proteinů – oxidace postranních amk zbytků, vznikají kroslinky (disulfidické vazby), fragmentace proteinů Vznik lézí na DNA – reakce s tyminem, vznik jednořetězcových zlomů Mechanismy pro odstranění ROS: Jsou nestabilní, rozpadají se spontáně, mnoho enzymatických a neenzymatických systémů jejich inaktivace v buňce Antioxidanty: blokují jejich tvorbu nebo je vychytávají (vit. A, E, C, glutathion) Transportní proteiny (laktoferrin, ferritin) vážou Fe, Hg ionty, snížení tvorby OH- Enzymy (kataláza, superoxiddismutáza, glutathion peoxidáza)
 poškození membrán a organel. ukončení vychytávačem volných radikálů (vitamin E) Oxidativní modifikace proteinů – oxidace postranních amk zbytků, vznikají kroslinky (disulfidické vazby), fragmentace proteinů. Vznik lézí na DNA – reakce s tyminem, vznik jednořetězcových zlomů. Mechanismy pro odstranění ROS: Jsou nestabilní, rozpadají se spontáně, mnoho enzymatických a neenzymatických systémů jejich inaktivace v buňce. Antioxidanty: blokují jejich tvorbu nebo je vychytávají (vit. A, E, C, glutathion) Transportní proteiny (laktoferrin, ferritin) vážou Fe, Hg ionty, snížení tvorby OH- Enzymy (kataláza, superoxiddismutáza, glutathion peoxidáza)")
21
Role ROS v buňce

22
Defekty permeability membrány
Rysem mnoha typů buněčného poškození MTCH membrána, PM, další membrány Toxiny, virové proteiny, ischemie… Biochemické mechanismy vedoucí poškození membrány Nefunkční MTCH snížení syntézy fosfolipidů a akumulace volných mastných kyselin Ztráta membránových fosfolipidů -aktivace fosfolipáz vlivem zvýšení Ca2+ v cytosolu Abnormality cytoskeletu- vlivem zvýšení Ca2+ v cytosolu aktivace proteáz porušení, odtržení cytoskeletu náchylnost PM k roztržrní ROS Poškozené lipidové částice – acyl karnitin, lysofosfolipidy, účinek jako detergenty, vmezeřují se do fosfolipidové dvojvrstvy a způsobují změny permeability ztráta osmotické rovnováhy, influx tekutin a iontů stejně jako ztráta proteinů, enzymů, RNA, ATP poškození lysozomů (enzymy RNázy, DNázy, proteázy, fosfatázy, katepsiny) enzymatické štěpení buněčných komponent … nekróza
 enzymatické štěpení buněčných komponent … nekróza.")
24
Reverzibilní a ireverzibilní buněčné poškození
Buněčné poškození snížená tvorba ATP, ztráta membránové integrity, defekty syntézy proteinů, poškození cytoskeletu, DNA Reverzibilní – vyrovnání s poškozením, po odeznění stresu zpět do normálního stavu Ireverzibilní – přetrvávající stres práh rozsáhlé poškození membrány, bobtnání lysosomů, vakuolizace MTCH, snížená schopnost syntézy ATP, ztráta proteinů, enzymů, RNA, metabolitů, permeabilní PM Molekulární mechanismy vedoucí k buněčné smrti ne všechny poškození jsou vždy fatální, ?rozdíl mezi primárním a sekundárním poškozením , chybí prahový bod, co je příčina a co důsledek… Kdy je buňka předurčena k smrti: Není schopná obnovit funkci MTCH (ox. fosforylace, ATP) Poškození membrán (únik proteinů, enzymů,metabolitů, zvýšení ic pH…)
 Poškození membrán (únik proteinů, enzymů,metabolitů, zvýšení ic pH…)")
25
Morfologie buň. poškození,Nekróza
Morfologické a biochemické změny nekróza Projevy efektů po různých dobách (minuty -reverzibilní, hodiny, dny) Reverzibilní poškození Bobtnání – ztráta funkce iontových pump v membráně ztráta iontové a osmotické homeostáze Změna metabolismu tuků – lipidové vakuoly PM – odškrcování měchýřků Bobtnání MTCH, rozředění Zvětšení ER, oddělení a rozpad polyzomů Změny jádra
 Reverzibilní poškození. Bobtnání – ztráta funkce iontových pump v membráně ztráta iontové a osmotické homeostáze. Změna metabolismu tuků – lipidové vakuoly. PM – odškrcování měchýřků. Bobtnání MTCH, rozředění. Zvětšení ER, oddělení a rozpad polyzomů. Změny jádra.")
26
Nekróza Spektrum morfologických změn v důsledku škodlivého působení enzymů Ztráta membránové integrity ztráta obsahu zánět Degradace proteinů, enzymatické štěpení buňky (lysozomy, cytosol) V cytoplasmě vakuoly, jsou v ní díry po naštěpených organelách, porušené membrány, rozšíření MTCH-amforní tělíska, jádro- aktivita DNáz, scvrknutí a fragmentace jádra Nekrotické buňky a jejich drť je odstraněna enzymatickou digescí, fragmentací a fagocyty, nedostatečné odstranění kalcifikace
 V cytoplasmě vakuoly, jsou v ní díry po naštěpených organelách, porušené membrány, rozšíření MTCH-amforní tělíska, jádro- aktivita DNáz, scvrknutí a fragmentace jádra. Nekrotické buňky a jejich drť je odstraněna enzymatickou digescí, fragmentací a fagocyty, nedostatečné odstranění kalcifikace.")
27
Příklady buněčného poškození vedoucí k nekróze
Ischemie a hypoxie - u ischemie dochází k nedostatku zásobení jak kyslíkem tak glukózou, akumulace metabolitů, jenž měly být odvedeny krví rychlejší poškození než u hypoxie ISCHEMIE: Snížené množství hemoglobinu, kolaps krevního tlaku, ztráta krve, infarkt Do určitého bodu se po obnovení krevního zásobení poškozená buňka navrací do normálu. Při překročení délky do prahového bodu buňka degraduje, komponenty glykolýzy a ox. fosforylace jsou nenávratně poškozeny a ani po obnovení zásobení nedojde k reverzi. Mechanismus : … + rozptýlení cytoskeletu, ztráta řasinek, tvorba měchýřků na povrchu buňky, bobtnání buňky a organel vratné změny Pokud ischemie přetrvává nevratné změny (masivní bobtnání, poruchy PM,MTCH-amforní tělíska, influx Ca2+) hlavně nekróza (x i apoptóza)
 hlavně nekróza (x i apoptóza)")
29
Příklady buněčného poškození vedoucí k nekróze
2. Ischemické reperfuzní poškození Reperfuze- obnovení zásobení krví - infarkt, mrtvice Reoxygenace zvýšení volných kyslíkových radikálů z buněk endotelu, parenchymu a leukocytů MTCH tvorba kanálků v membráně zánět, infiltrace neutrofilů 3. Chemické poškození Přímá reakce s buněčnými komponentami (Hg se váže na sulfhydrylové skupiny zvýšená permeabilita PM, inhibice transportu, kyanid blokuje ox. fosforylaci) Chemikálie jsou biologicky aktivní až po metabolizaci buňkou (P-450) CCl CCl3 (CCl4 + e CCl3 + Cl-) volné radikály způsobují autooxidaci mastných kyselin peroxidace lipidů (vznik dalších ROS),poškození ER v důsledku rozpadu lipidů, oddělení polyzomů od HER, poškození PM, influx Na+, H2O, Ca2+, bobtnání aktivace enzymů smrt
 Chemikálie jsou biologicky aktivní až po metabolizaci buňkou (P-450) CCl4 CCl3 (CCl4 + e- CCl3 + Cl-) volné radikály způsobují autooxidaci mastných kyselin peroxidace lipidů (vznik dalších ROS),poškození ER v důsledku rozpadu lipidů, oddělení polyzomů od HER, poškození PM, influx Na+, H2O, Ca2+, bobtnání aktivace enzymů smrt.")
31
Subcelulární odpověď na poškození
Katabolismus lysozomů Membránově vázané organely – hydrolytické enzymy (odbourání proteinů,nukleových kyselin, oligosacharidy, fosfolipidy) Odstranění fagocytovaného materiálu heterofagií nebo autofagií HETEROFAGIE Lysosomální štěpení extracelulárního materiálu endocytóza Fagocytóza, pinocytóza Vakuoly (endozomy, fagozomy) splývají s lysozomy fagolysozomy štěpení matriálu Neutrofily, makrofágy
 Odstranění fagocytovaného materiálu heterofagií nebo autofagií. HETEROFAGIE. Lysosomální štěpení extracelulárního materiálu endocytóza. Fagocytóza, pinocytóza. Vakuoly (endozomy, fagozomy) splývají s lysozomy fagolysozomy štěpení matriálu. Neutrofily, makrofágy.")
32
2. HYPERTROFIE HLADKÉHO ER
AUTOFAGIE Lysosomální digesce vlastních buněčných komponent Cytoplasma autofagní vakuola (obsahující buněčnou komponentu) fúze s lysozomy nebo s elementy GA vznik autofagolysozom štěpení Běžný jev při odstráňování poškozených buněčných organel, remodelaci a diferenciaci, často u atrofických buněk Enzymy štěpí proteiny a uhlovodíky,ale některé lipidy digesci nepodléhají a tvoří reziduální tělíska nebo jsou vypuštěny ven z buňky. Neštěpené lipidy tvoří pigment lipofuscin pigment z tetování zůstává ve fagolysozomech desítky let Poruchy lysozomů (deficience některých enzymů nebo v důsledku chemických látek) hromadění obsahu lysozomů 2. HYPERTROFIE HLADKÉHO ER Adaptace na léčiva- potřeba vyšších dávek v důsledku zvětšení ER hepatocytů metabolizace látek přeměna P 450 v ER na rozpustnější formy, umožňující jejich sekreci x větší toxicita+tvorba ROS Látky stimulují syntézu těchto enzymů a hladkého ER
 fúze s lysozomy nebo s elementy GA vznik autofagolysozom štěpení. Běžný jev při odstráňování poškozených buněčných organel, remodelaci a diferenciaci, často u atrofických buněk. Enzymy štěpí proteiny a uhlovodíky,ale některé lipidy digesci nepodléhají a tvoří reziduální tělíska nebo jsou vypuštěny ven z buňky. Neštěpené lipidy tvoří pigment lipofuscin. pigment z tetování zůstává ve fagolysozomech desítky let. Poruchy lysozomů (deficience některých enzymů nebo v důsledku chemických látek) hromadění obsahu lysozomů. 2. HYPERTROFIE HLADKÉHO ER. Adaptace na léčiva- potřeba vyšších dávek v důsledku zvětšení ER hepatocytů metabolizace látek přeměna P 450 v ER na rozpustnější formy, umožňující jejich sekreci x větší toxicita+tvorba ROS. Látky stimulují syntézu těchto enzymů a hladkého ER.")
33
3. ZMĚNY MTCH -změny počtu, velikosti, tvaru -Hypertrofie, atrofie- zvýšení resp. snížení počtu mtch Megamitochondrie – játra alkoholika, podvýživa Mitochondriální myopatie:zvýšený počet mtch s abnormálními kristami a krystaliny

34
4. ABNORMALITY CYTOSKELETU
mikrotubuly, aktinová filamenta, myosinová filamenta a intermediální filamenta, kontraktilní proteiny architektura buňky, pohyb, adheze Poruchy transferu komponent, pohybu organel.. Mikrofilamenta (aktin, myosin,asociované proteiny) - pohyb leukocytů, fagocytů - Cytochalasin B zabraňuje polymerizaci aktinu, phalloidin váže aktinové filamenta Mikrotubuly (tubulin) - defekty –pohyb spermií infertilita, pohyb leukocytů, fagocytů - kolchicin se váže na tubulin, vinca alkaloidy se vážou na mikrotubuly (protinádorový účinek) Intermediální filamenta - keratinová filamenta, neurofilamenta, desminy, vimentiny, gliová filamenta - myopatie, neurologické poruchy, nemoci kůže
 - pohyb leukocytů, fagocytů. - Cytochalasin B zabraňuje polymerizaci aktinu, phalloidin váže aktinové filamenta. Mikrotubuly (tubulin) - defekty –pohyb spermií infertilita, pohyb leukocytů, fagocytů. - kolchicin se váže na tubulin, vinca alkaloidy se vážou na mikrotubuly (protinádorový účinek) Intermediální filamenta. - keratinová filamenta, neurofilamenta, desminy, vimentiny, gliová filamenta. - myopatie, neurologické poruchy, nemoci kůže.")
35
Intracelulární ukládání
Jedním z důsledků poruch buněčného metabolismu je nadměrné ukládání buněčných komponent Buňce vlastní komponenty (voda, lipidy, uhlovodíky, proteiny) Buňce cizí substance (exogenní agens, infekce, abnormální metabolity) Pigment dočasně, permanentně, poškozující až toxické, v cytoplazmě nebo v jádře 3 typy: Buňce vlastní substance je syntetizována v normální nebo vyšší míře,ale metabolismus není schopný ji odbourat Buňce vlastní substance či abnormální substance není odbourávána kvůli genetické poruše defektní metabolismus, transport, skládání nebo sekrece substance (defekty enzymů -nejsou schopny odbourat jejich přirozený substrát) Abnormální substance je ukládána, neboť buňka pro ni nemá enzymatický ani transportní systém
 Buňce cizí substance (exogenní agens, infekce, abnormální metabolity) Pigment. dočasně, permanentně, poškozující až toxické, v cytoplazmě nebo v jádře. 3 typy: Buňce vlastní substance je syntetizována v normální nebo vyšší míře,ale metabolismus není schopný ji odbourat. Buňce vlastní substance či abnormální substance není odbourávána kvůli genetické poruše defektní metabolismus, transport, skládání nebo sekrece substance (defekty enzymů -nejsou schopny odbourat jejich přirozený substrát) Abnormální substance je ukládána, neboť buňka pro ni nemá enzymatický ani transportní systém.")
36
Ukládání lipidů Tryglyceridy, cholesterol/estery, fosfolipidy…
Ukládání fosfolipidů a myelinových útvarů nekróza Steatóza – nahromadění triglyceridů v buňkách parenchymu (játra, srdce, svaly, ledviny) toxiny, diabetes mellitus, obezita, anoxie, alkohol Různé mechanismy způsobující hromadění tryglyceridů v játrech Mastné kyseliny metabolizace v hepatocytech porucha v jakékoliv části této dráhy Ukládání tuku v játrech (3-6 kg) - malé vakuolky kolem jádra splynutí vytlačení jádra na okraj buňky Srdce – ukládání tuku v důsledku hypoxie
 toxiny, diabetes mellitus, obezita, anoxie, alkohol. Různé mechanismy způsobující hromadění tryglyceridů v játrech. Mastné kyseliny metabolizace v hepatocytech. porucha v jakékoliv části této dráhy. Ukládání tuku v játrech (3-6 kg) - malé vakuolky kolem jádra splynutí. vytlačení jádra na okraj buňky. Srdce – ukládání tuku v důsledku hypoxie.")
37
Cholesterol a cholesterolové estery
Využití pro syntézu membrán bez patologické akumulace Hromadění ve vakuolách při: Ateroskleróza – ukládání cholesteru v buňkách hladkého svalstva a makrofágů v aortě a větších tepnách Xantom – hromadění cholesterolu v makrofázích tukové bulky v podkoží Zánět a nekróza- makrofágy v místě zánětu a poranění, fagocytují zbytky membrán (cholesterol) Cholesteróza – ukládání v žlučníku
 Cholesteróza – ukládání v žlučníku.")
38
Ukládání proteinů Kulaté kapičky, vakuoly nebo agregáty v cytoplasmě
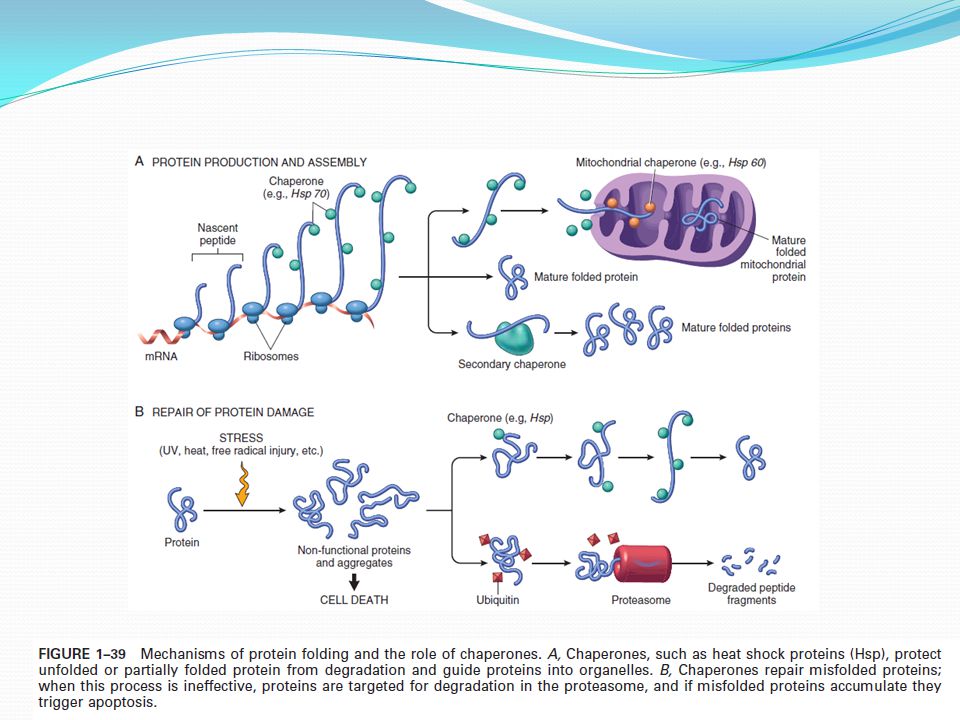
Příčiny: Proteinurie – v ledvinovém epitelu zvýšená reabsorbce proteinů Nadměrná syntéza proteinu- při syntéze Ig zvětšení ER Russelovy tělíska Špatné skládání proteinů - nascentní polypeptidový řetězec je sestavován do konformace α-helixu nebo β -listu funkce a transport - částečně složené proteiny se můžou shlukovat nebo se zamotat s jinými proteiny - tyto intermediáty jsou stabilizovány chaperony- napomáhají skládání a transportu proteinů - některé jsou syntetizovány konstitutivně a jiné jsou indukovány stresem (teplem –HSP) správné skládání x degradace poškozeného proteinu (ubikvitin proteazomová)
 správné skládání x degradace poškozeného proteinu (ubikvitin proteazomová)")
39
Špatné skládání proteinů a jejich hromadění v buňce:
Defektní transport a sekrece proteinu (cystická fibróza, disociace proteinu chloridového kanálu od chaperonu) ER stres - špatně složené proteiny se hromadí v ER a spouštějí odpověď na ně (unfolded protein response) - spuštěná různými proteiny roztažení ER aktivace signálních drah redukce špatně složených proteinů zvýšení produkce chaperonů a snížení translace proteinů - spouští apoptózu kaspáza 12 (ER) - neurodegenerativní choroby Alzheimer, Parkinson, Huntington - Agregace abnormálních proteinů – ukládání v tkáních extracelulárně i intracelulárně, interferují s normální funkcí (amyloidóza)
 ER stres - špatně složené proteiny se hromadí v ER a spouštějí odpověď na ně (unfolded protein response) - spuštěná různými proteiny roztažení ER aktivace signálních drah redukce špatně složených proteinů zvýšení produkce chaperonů a snížení translace proteinů. - spouští apoptózu kaspáza 12 (ER) - neurodegenerativní choroby Alzheimer, Parkinson, Huntington. - Agregace abnormálních proteinů – ukládání v tkáních extracelulárně i intracelulárně, interferují s normální funkcí (amyloidóza)")
41
Hyalin Intracelulární: histologické označení mnoha typů hromadění proteinů (Russelova tělíska, Malloryho tělíska (šipka) způsobená alkoholem v hepatocytech) Extracelulární Polysacharid- zásoba energie v cytoplazmě Zvýšené ukládání při poruše metabolismu glukózy či glykogenu Vakuoly v cytoplazmě Diabetes mellitus a jiné genetické choroby (glykogen storage diseases -defekty enzymů) Glykogen
 způsobená alkoholem v hepatocytech) Extracelulární. Polysacharid- zásoba energie v cytoplazmě. Zvýšené ukládání při poruše metabolismu glukózy či glykogenu. Vakuoly v cytoplazmě. Diabetes mellitus a jiné genetické choroby (glykogen storage diseases -defekty enzymů) Glykogen.")
42
Pigment Barevné substance ukládané v buňce, exogení, endogení, přirozené(melanin)x cizorodé Exogení- uhlík, uhelný prach- znečištění prostředí inhalace makrofágy lymfatické uzliny průdušky, průdušnice usazování, zčernání plic a uzliny - pracující v dolech rozedma plic - tetování pigment je fagocytován kožními makrofágy, kde zůstávají Endogení - Lipofuscin (hnědý nerozpustný pigment, pigment stárnutí) - polymer lipidů a fosfolipidů+ proteiny, poukazuje na oxidativní stres a lipidovou peroxidaci - u buněk s pomalými regresivními změnami - během stárnutí – játra, srdce Melanin – hnědočerný, melanocyty, nadměrně se ukládá u alkaptonurie
 - polymer lipidů a fosfolipidů+ proteiny, poukazuje na oxidativní stres a lipidovou peroxidaci. - u buněk s pomalými regresivními změnami. - během stárnutí – játra, srdce. Melanin – hnědočerný, melanocyty, nadměrně se ukládá u alkaptonurie.")
43
zlato-žluto-hnědý pigment, Fe
Hemosiderin zlato-žluto-hnědý pigment, Fe Při nadbytku železa ferritin tvoří hemosiderinové granula Hemosideróza -nadměrné množstí železa -akumulace ve fagocytech (makrofázích) popř. buňkách parenchymu Při: nadbytku železa v potravě nedostatečné zpracování železa anémie transfuze - nepoškozuje buňky parenchymu (x hemochromatóza-chronické zatížení železem, ne jen dočasné, „koroze“ orgánů- poškození jater, srdce, slinivky)
 popř. buňkách parenchymu Při: nadbytku železa v potravě. nedostatečné zpracování železa. anémie. transfuze. - nepoškozuje buňky parenchymu (x hemochromatóza-chronické zatížení železem, ne jen dočasné, „koroze orgánů- poškození jater, srdce, slinivky)")
44
Patologická kalcifikace
Nadměrné ukládání vápenatých solí, s malým množství Fe, Mg a jiných minerálních solí Distrofycká V odumírajících tkáních, za fyziologických hodnot Ca, porucha metabolismu Ca Nekróza, ateroskleróza, při stárnutí, poškození srdečních chlopní Malé bíle granulky nebo shluky zrníček Způsobuje nefunkčnost orgánů Metastatická V normálních tkáních při zvýšeném množství Ca (hyperkalcémie) -zvýšená hladina hormonů příštítných tělísek(tumor příštítných tělísek nebo jiný tumor sekretující hormony) - poranění kosti (tumory kostní dřeně - mnohočetný myelom, leukemie;metastáze do kostí) - nadměrný příjem vitaminu D - poruchy ledvin sekundární nadměrná tvorba hormonů příštítných tělísek - ledviny, plíce, tepny, plicní žíly, zažívací trakt
 -zvýšená hladina hormonů příštítných tělísek(tumor. příštítných tělísek nebo jiný tumor sekretující hormony) - poranění kosti (tumory kostní dřeně - mnohočetný myelom, leukemie;metastáze do kostí) - nadměrný příjem vitaminu D. - poruchy ledvin sekundární nadměrná tvorba hormonů příštítných tělísek. - ledviny, plíce, tepny, plicní žíly, zažívací trakt.")
45
Buněčné stárnutí Fyziologické a strukturální změny orgánů během stárnutí Ovlivněno genetickými faktory, environmentálními faktory, nemocemi Progresivní hromadění subletálního poškození snížená schopnost odpovídat na stres a poškození, buněčná smrt

46
Strukturální a biochemické změny buněčného stárnutí
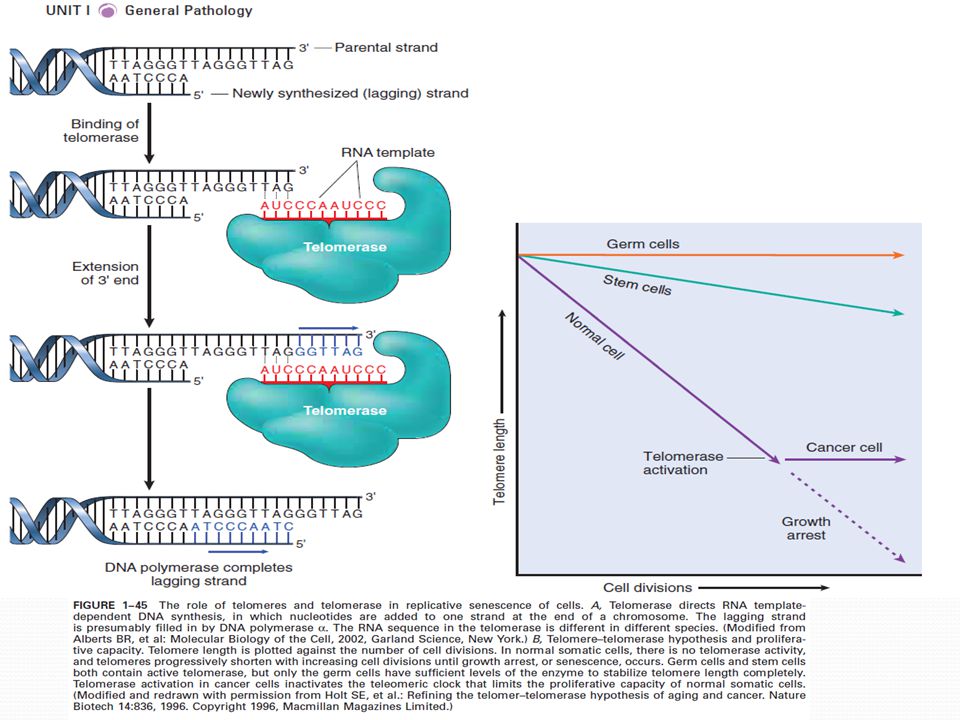
Redukce ox. fosforylace , syntézy nukleových kyselin, strukturálních a enzymatických proteinů, receptorů a TF Snížená kapacita příjmu živin Snížená schopnost opravy DNA Morfologické změny: abnormálně laločnaté jádro, mnohotvaré MTCH, zmenšení ER, deformovaný GA, lipofusin –produkt peroxidace lipidů, důsledek oxidativního stresu Omezený počet dělení, pak se buňky dostávají do konečného nedělícího stádia SENESCENCE Změny genové exprese (přesně řízený proces), inhibitory CDK (p21) jsou overexprimovány Mechanismus je stále zkoumán při každé replikaci nedokončená replikace konců chromozomů (zkrácení telomer) zastavení BC Telomery : tandemové repetice na koncích chromozomů TTAGGG, důležité pro zajištění kompletní replikaci konců chromozomů Při replikaci je malá část telomer nedoreplikována zkrácení telomer enzym telomeráza- specializovaný enzymový komplex s RNA, kterou používá jako templát pro přidání nukleotidů na konec chromozomu V zárodečných a kmenových buňkách, ne v somatických stárnutí (x rakovinné buňky) Replikativní potenciál buněčného stárnutí
, inhibitory CDK (p21) jsou overexprimovány. Mechanismus je stále zkoumán při každé replikaci nedokončená replikace konců chromozomů (zkrácení telomer) zastavení BC. Telomery : tandemové repetice na koncích chromozomů TTAGGG, důležité pro zajištění kompletní replikaci konců chromozomů. Při replikaci je malá část telomer nedoreplikována zkrácení telomer enzym telomeráza- specializovaný enzymový komplex s RNA, kterou používá jako templát pro přidání nukleotidů na konec chromozomu. V zárodečných a kmenových buňkách, ne v somatických stárnutí (x rakovinné buňky) Replikativní potenciál buněčného stárnutí.")
48
Hromadění metabolických a genetických změn
Stárnutí může být podmíněno buněčným poškozením vyplývajícím z metabolických událostí Menší zvířata kratší život rychlejší metabolismus Množství oxidativního stresu ROS, lipofuscin Délka života koreluje se zvýšeným množstvím SOD a sníženým množstvím SO Poškození DNA oprava opravnými systémy buňky x některé se hromadí Wernerův syndrom (porucha helikázy), Ataxia telangiectasia(porucha opravného mechanismu)
, Ataxia telangiectasia(porucha opravného mechanismu)")
49
Dotazy:g. rylova@gmail
Studijní matriály: Robbins pathology Chapter 1:Cellular Adaptations, Cell Injury, and Cell Death (kapitola volně stažitelná z internetu)
")
Podobné prezentace