Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
VÝVOJ NOVÉHO LÉČIVA

2
Drug development 0,8 – 1 mld. USD

3
Objev nového léčiva objevování nových vlastností známých chemických látek modifikací chemické struktury již známého léčiva využití přírodních látek cílená syntéza látek – lékový design

4
Preklinické testování

5
Preklinické modely in silico - QSAR – Predikce/simulace (na základě fyzikálně chemických vlastností) in vitro – izolované orgány, tkáňové proužky, buňky, subcelulární částice (mikrozomy, exprimované enzymy) in vivo - laboratorní zvířata
 in vitro – izolované orgány, tkáňové proužky, buňky, subcelulární částice (mikrozomy, exprimované enzymy) in vivo - laboratorní zvířata")
6
Cíle preklinického testování PD účinek – „Proof of Principle“ – nejdříve – farmakologický screening PK profil Identifikovat potenciální nežádoucí účinky/interakce/toxicity – Cílové orgány toxicity – Reverzibilita změn Stanovit podmínky bezpečného podání u lidí – Určit bezpečné a „účinné“ počáteční dávky – Stanovit hlavní monitorovací kritéria pro klinické studie Dodat data, že lidé nebudou vystavení nepřiměřenému riziku – Zároveň předpoklad jisté účinnosti

7
Preklinické studie bezpečnosti Bezpečnostní farmakologie – orgánová toxicita Toxikokinetika/farmakokinetika Akutní toxicita Chronická toxicita Speciální studie toxicity – Karcinogenicita – Mutagenicita – Genotoxicita – Fototoxicita – Lokální tolerance – Spojivkový vak – oko – Reprodukční toxicita

8
Bezpečnostní farmakologie Zhodnocení vlivu látky na specifické orgánové systémy Nejčastěji – Kardiovaskulární (QT interval…) – CNS (konvulze, sedace atd.) – Respirační systém Mohou být provedeny zvlášť či jako součást chronické toxicity
 – CNS (konvulze, sedace atd.) – Respirační systém Mohou být provedeny zvlášť či jako součást chronické toxicity")
9
Toxikokinetika & farmakokinetika Vývoj analytické metody Testování prvotních fází vývoje lékové formy Testování různých dávek a dávkovacích schémat ADME u různých druhů zvířat Preklinická PK/PD - vztah k bezpečnosti

10
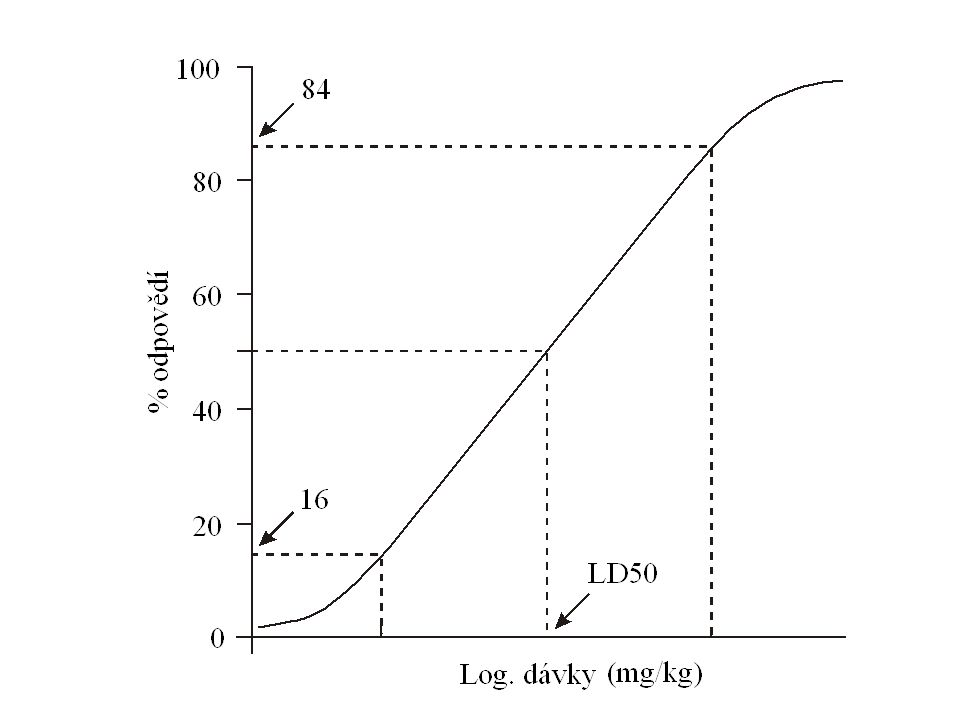
Zkoušky na toxicitu (bezpečnost) GLP studie Akutní toxicita – u dvou druhů savců - nejčastěji potkan a pes pro malé molekuly – primáti pro biologické látky – Umožňuje odhad dávek pro další studie Chronická toxicita – Vychází se z předpokládané délky trvání klinického použití Taky z předpokládaného dávkovacího schématu, cesty podání a lékové formy – jednorázově -------- jednotýdenní – po dobu 1 týdně ---- jednoměsíční – po dobu 1 měsíce -- tříměsíční – > měsíc ------- > 6ti měsíční
 GLP studie Akutní toxicita – u dvou druhů savců - nejčastěji potkan a pes pro malé molekuly – primáti pro biologické látky – Umožňuje odhad dávek pro další studie Chronická toxicita – Vychází se z předpokládané délky trvání klinického použití Taky z předpokládaného dávkovacího schématu, cesty podání a lékové formy – jednorázově jednotýdenní – po dobu 1 týdně ---- jednoměsíční – po dobu 1 měsíce -- tříměsíční – > měsíc > 6ti měsíční")
15
Rozhodnutí podat první dávku člověku povoluje SÚKL, – jestliže nová látka přináší výhody ve srovnání s farmakoterapeutickými postupy založenými na využití současných léčivých přípravků Klíčové varujícící informace (ve vztahu k dávce): – kinetika – nelineární kinetika, genetický polymorfizmus, nízká clearance... – dynamika (benefit vs. rizika) neznámý účinek na cílovou strukturu, významný vliv na životně důležité funkce, stimulace buněčné proliferace... – toxicita nečekané úhyny
 neznámý účinek na cílovou strukturu, významný vliv na životně důležité funkce, stimulace buněčné proliferace... – toxicita nečekané úhyny.")
16
Klinické hodnocení

17
jakékoli systematické testování prováděné na subjektech hodnocení za účelem – 1. zjistit či ověřit klinické, farmakologické nebo jiné – farmakodynamické účinky, – 2. stanovit nežádoucí účinky – 3. studovat absorpci, distribuci, metabolismus nebo vylučování jednoho nebo několika hodnocených LP s cílem ověřit bezpečnost nebo účinnost tohoto LP nebo LPs, včetně KH probíhajících v jednom nebo v několika místech KH v ČR, popřípadně v dalších členských státech zákon 378/2007 Sb. § 51 odst. 2 písm. a)
.")
18
Správná klinická praxe je soubor mezinárodně uznávaných etických a vědeckých požadavků na jakost, – které musí být dodržovány při navrhování, provádění, zaznamenávání a předkládání zpráv o klinických hodnoceních s účastí lidských subjektů. Direktiva 2001/20/EK

19
Klinické hodnocení Fáze I - první podání člověku Fáze II – terapeutická - podání léčiva již v předpokládané indikaci pacientům – Fáze IIa, kdy dochází k hodnocení předpokládaných indikací a k vyhledávání nežádoucích účinků – Fáze IIb, jež zahrnuje některé dílčí studie zaměřené na stanovení farmakodynamiky, farmakokinetiky a biotransformace při opakovaném podávání v základních indikacích. Fáze III - Rozšířená klinická studie Fáze IV (postregistrační, postmarketingové hodnocení)
.")
20
I. Fáze - první vyzkoušení LP na člověku předběžná klinická studie na zdravých dobrovolnících – i nevyléčitelně nemocných - cytostatika Cíl - zjistit bezpečnost LP podáním – malé dávky - obvykle < 1/10 dávky v mg/kg, která nevyvolala toxické účinky u zvířete – dávka se zvyšuje, opakuje, sledují se NÚ (dynamika), kinetika 50-100 subjektů
, kinetika subjektů.")
21
2. Fáze - orientační klinický pokus ověření bezpečnosti LP u nemocného člověka – Úzce vymezený okruh subjektů se stejným onemocněním zkoumá se rozsah terapeutických dávek a cesta podání doplňovány informace o kinetice a dynamice za patologického stavu 100-300 subjektů

22
Fáze III - Rozšířená klinická studie – Také kontrolovaný klinický pokus kritické zhodnocení farmakoterapeutického účinku - zásady: – kontrolní skupina (placebo, referenční LP) – zkřížený pokus s vymývací periodou (washout) – zaslepení studie – randomizace, pečlivý výběr probandů podle předem stanovených kritérií (stadium nemoci, hmotnost, věk, pohlaví) poznatky: indikace a kind dokumenty: příbalová informace nemocného. SPC 1000 – 5000 subjektů – „populace“

23
REGISTRACE znamená zařazení nového LP pro danou LF, dávky a indikace do registru LP povolených k užívání v daném území (státu). V ČR uděluje SÚKL. – Žádá o ni výrobce LP, je povinen předložit požadovanou dokumentaci o terapeutických účincích a riziku. Platí 5 let, – poté je třeba požádat o obnovení registrace.

24
4. FÁZE: postmarketingová po dobu 4-5 let k ověření nového LP v širší praxi z hlediska bezpečnosti. – Definitivní rozhodnutí, do jaké míry je LP účinný a bezpečný LP v rukou široké populace s nejrůznějšími interindividuálními odchylkami v odpovědi na LP projeví se NU s nízkou incidencí – K tomu účelu slouží: monitorování NU – formulář: “Zpráva o nežádoucím účinku LP“ – Národní centrum pro sledování NU LP, v ČR SÚKL a příslušná komise -databáze - WHO oprávněnost indikací a kind

25
Souhrnná informace o LP (SPC) kompletní a závazná informace o LP předkládá výrobce – na základě dat z preklinického a klinického hodnocení LP schvaluje SÚKL databáze: AISLP, databáze SÚKLu
 kompletní a závazná informace o LP předkládá výrobce – na základě dat z preklinického a klinického hodnocení LP schvaluje SÚKL databáze: AISLP, databáze SÚKLu")
Podobné prezentace




![[ 1 ] RNDr. Pavla Coufalová Symposium Strategie servisní podpory zdravotnických pracovišť, Praha, 7. 10. 2010 © 2010 Státní ústav pro kontrolu léčiv.](/41/11203357/big_thumb.jpg "[ 1 ] RNDr. Pavla Coufalová Symposium Strategie servisní podpory zdravotnických pracovišť, Praha, 7. 10. 2010 © 2010 Státní ústav pro kontrolu léčiv.>")


![[ 1 ] MUDr. Jana Mladá Farmakovigilance © 2010 Státní ústav pro kontrolu léčiv 5 minut pro bezpečnější farmakoterapii – proč se zabývat nežádoucími účinky.](/42/11301027/big_thumb.jpg "[ 1 ] MUDr. Jana Mladá Farmakovigilance © 2010 Státní ústav pro kontrolu léčiv 5 minut pro bezpečnější farmakoterapii – proč se zabývat nežádoucími účinky.>")













