Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
NAŘÍZENÍ PRO KH A DALŠÍ DŮLEŽITÉ INFORMACE Sekce registrací Oddělení klinického hodnocení Oddělení posuzování farmaceutické dokumentace 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 1

2
SÚKL – ředitel MUDr. Zdeněk Blahuta vedoucí kanceláře ředitele – Mgr. Klára Zachová zrušeny pozice náměstků + VUKOČ vedoucí Sekce registrací – MUDr. Jana Mladá PPR – právní podpora registrací Mgr. Kateřina Blechová Mgr. Eliška Járosová Mgr. Lucie Bulandrová Mgr. Karel Ulrych plné moci – PRO – Mgr. Michal Paris © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 2 Organizační a personální změny 3. 6. 2014

3
odd. KH – posíleno MUDr. Terézie Prosbová MUDr. Eva Hrušková Reinová MUDr. Jana Kratoňová MUDr. Ondřej Palán Ing. Eva Kolouchová, Ph.D. – preklinika Mgr. Markéta Čermáková – koordinátor © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 3 Oddělení KH + FD 3. 6. 2014

4
chemické LP Ing. Tereza Stefflová Mgr. Jiřina Humpolíková Mgr. Helena Seidlová biotechnologicky vyráběné LP Ing. Lucie Zmatlíková (LPMT) Ing. Ivana Haunerová (LPMT; CAT) RNDr. Lenka Břízová, Ph.D. Ing. Kateřina Pospíšilová (vakcíny) Ing. Dagmar Pospíšilová 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 4 Oddělení FD
 Ing. Ivana Haunerová (LPMT; CAT) RNDr. Lenka Břízová, Ph.D. Ing. Kateřina Pospíšilová (vakcíny) Ing. Dagmar Pospíšilová © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 4 Oddělení FD.")
5
Program semináře: Zahájení Regulation of the European Parlament and of the Council in clinical trials on medicinal products for human use and repealing Directive 2001/20/EC (A.Němcová) přestávka Sdělení výsledků dizertační práce PharmDr.E.Gruberové na téma “Informace pro pacienty/Informované souhlasy“ (E.Gruber) Pokyn k Informacím pro subjekty hodnocení/informovaným souhlasům“ (A.Trunečková) Přestávka Nejčastější chyby a nedostatky při předkládání žádostí o KH a v průběhu schvalování (L.Kraváčková) Opakující se nedostatky v předkládané farmaceutické dokumentaci (T.Stefflová) Nedostatky zasílaných zpráv a informací v průběhu KH (T.Boráň) 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 5

6
Nařízení pro KH a předpokládaný dopad změn MUDr. Alice Němcová Sekce registrací Oddělení klinického hodnocení 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 6

7
Directive 2001/20/EC of the European Parliament and of the Council on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use 4. dubna 2001 do 1. května 2004 implementována do národní legislativy 2007 – 1. veřejná diskuse 2010 – 2. veřejná diskuse Po celou dobu – sledování implementace, harmonizace a podnětů k aktualizaci směrnice 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 7

8
Proposal for a Regulation of the European Parlament and of the Council in clinical trials on medicinal products for human use and repealing Directive 2001/20/EC Návrh předložen v červenci 2012 Projednáván v Radě EU a EU parlamentu Schválen v Radě EU koncem roku 2013 Schválen EU parlamentem – 2.4.2014 ± ½ 2014 – schválení EC – vydání ve Věstníku EC Platnost – musí být funkční Portál a databáze KH Bude platný nejen v EU, ale i v EHS 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 8

9
NAŘÍZENÍ EVROPSKÉHO PARLAMENTU A RADY (EU) č. 536/2014 ze dne 16. dubna 2014 o klinických hodnoceních humánních léčivých přípravků a o zrušení směrnice 2001/20/ES REGULATION (EU) No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC Zveřejněn ve Věstníku EC 27. 5. 2014 Vstup v platnost 16. 6. 2014 (20.den po zveřejnění) Nabytí účinnosti 6 měsíců po zveřejnění info o funkčnosti Portálu a EU databáze KH, ne dříve než 28. 5. 2016 (a déle) © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 9
 No 536/2014 OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 16 April 2014 on clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC Zveřejněn ve Věstníku EC Vstup v platnost (20.den po zveřejnění) Nabytí účinnosti 6 měsíců po zveřejnění info o funkčnosti Portálu a EU databáze KH, ne dříve než (a déle) © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 9.")
10
celé znění: http://eur-lex.europa.eu/legal- content/CS/TXT/?uri=OJ:L:2014:158:TOC http://eur-lex.europa.eu/legal- content/CS/TXT/?uri=OJ:L:2014:158:TOC 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 10

11
Zdůvodnění návrhu nařízení Zjednodušení administrativy pro zadavatele (i akademický výzkum) Zastavit pokles předkládaných žádostí v EU - od roku 2007 – 2010 pokles o 25 % Zrychlení posuzovacího procesu multinational CTAs Zatraktivnění ČS EU pro zadavatele All CTAs 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 11

12
Portál Vytvoří EMA za spolupráce ČS Jednotný pro všechna KH Spojen s novou databází KH Veškerá komunikace se zadavatelem – prostřednictvím portálu Jedno kontaktní místo v ČS (SÚKL) Předkládání dokumentace prostřednictvím portálu Assessment report (AR), připomínky, inspekční zprávy, informace o zahájení, průběžné zprávy - vkládáno přes portál 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 12

13
Nová databáze Zřídí EMA, bude vést databázi Napojena na portál Propojení s EudraCT databází (?), EudraVigilance databází - bude funkčí i nadále Přístupná veřejnosti Souhrn o KH pro RA, EK, EMA, EC Souhrn o KH pro laiky Zajistit snadné vyhledávání Jednotný zvolený jazyk (angl.?) 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 13

14
Obecné požadavky Zdůraznění – ochrana SH a zajištění robustních dat z KH Zajištění lidských práv a důstojnosti člověka – dle Listiny základních lidských práv Pro KH – stále platí ICH E6 (zásady GCP) a Helsinská deklarace Dle legislativy či nastavení ČS Nastavená vlastního procesu posouzení CTA v ČS Zapojení etických komisí (ČS odpovědný za dodržování lhůt i EK) Přiměřený počet + nejméně 1 laik či pacient zapojeni do posuzování Bez střetu zájmů Nezávislé na zadavateli, instituci, kde bude KH probíhat, zkoušejícím, osobách financujících KH Nesmí mít osobní či finanční zájem Zdůrazněna – kvalifikace, zkušenosti Zranitelné SH – odborníci v oboru 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 14

15
Obecné požadavky Ochrana osobních údajů Transparentnost – zveřejňování dat z KH Dle národní legislativy Možnost užití abortiv v KH Lidských a zvířecích bb. v KH Posuzování KH – pružnější, rychlejší, nejsou stanoveny min. časy, jsou dány max. časy Podpora výzkumu – zjednodušení pro akademický výzkum (např. zjednodušená dokumentace, bez poplatku, zjednodušení značení, pojištění (?)…) 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 15 Návrh nařízení – hlavní obsah
…) © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 15 Návrh nařízení – hlavní obsah.")
16
Etické komise – nezávislý orgán v ČS ustavený dle národní legislativy a pověřený vydávat stanoviska ke KH – za účasti nezávislých osob, pacientů či pacientských organizací Zadavatel – může být i více zadavatelů – stanoveny kompetence a odpovědnost Legal Representative – zákonný zástupce sponzora Dle rozhodnutí ČS – postačující i usazení kontaktní osoby ńa území EU Legally designated representative – zákonně ustanovený zástupce (rodič či zákonný zástupce nezletilé či nesvéprávné osoby) Zahájení KH – první akt učiněný k náboru SH 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 16 Definice

17
Non-interventional study – se rozumí klinická studie, která není klinickým hodnocením Intervence nemá definici Normal Clinical Practice = běžnou klinickou praxí – léčebné režimy obvykle používané k léčbě, prevenci nebo diagnostice onemocnění či choroby IMP – investigational medicinal product = hodnocený LP (testovaný, srovnávací, placebo) AMP – auxiliary medicinal product = pomocné LP – pomocný LP používaný pro KH dle protokolu, ale nejde o IMP Významná změna (místo podstatného dodatku) 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 17 Definice

18
Odkaz některých definic na Směrnici 2001/83/EC (např. léčivý přípravek, radiofarmaka, nežádoucí účinek, závažný nežádoucí účinek, vnitřní obal a vnější obal) Nové rozdělení studií: Clinical Study = klinická studie Clinical Trial = klinické hodnocení Low-intervention clinical Trial = nízkointervenční klinické hodnocení Cluster study – není v definicích, přesto se v textu u informovaných souhlasů uvádí i s upřesněním, o jaké studie jde 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 18 Návrh nařízení – hlavní obsah
 Nové rozdělení studií: Clinical Study = klinická studie Clinical Trial = klinické hodnocení Low-intervention clinical Trial = nízkointervenční klinické hodnocení Cluster study – není v definicích, přesto se v textu u informovaných souhlasů uvádí i s upřesněním, o jaké studie jde © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 18 Návrh nařízení – hlavní obsah.")
19
Clinical study = klinická studie Jakékoli testování prováděné na lidech za účelem a)zjistit nebo ověřit klinické, farmakologické či farmakodynamické účinky jednoho nebo více LP b)zjistit nežádoucí účinky jednoho či více LP c)studium absorpce, distribuce, metabolismu a vylučování jednoho nebo více LP s cílem ověřit bezpečnost nebo účinnost těchto léčivých přípravků 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 19 Nové rozdělení studií – nové definice

20
Clinical trial = klinické hodnocení klinickým hodnocením se rozumí klinická studie, která splňuje některou z následujících podmínek: a)přiřazení SH ke konkrétní léčbě je dáno Protokolem a nespadá do běžné klinické praxe dotčeného ČS; b)rozhodnutí předepsat či podat hodnocené LP je společně s rozhodnutím zařadit SH do KH; nebo c)použité diagnostické nebo monitorovací postupy jsou nad rámec běžné klinické praxe 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 20 Nové rozdělení studií – nové definice

21
Low-intervention clinical trial nízkointervenční klinické hodnocení musí splňovat všechny následující podmínky: a)LP jsou registrovány (vyjma placeba) v některém ČS EU b)LP jsou používány dle Protokolem v souladu s rozhodnutímmo registraci nebo je jejich použití v souladu se standardní léčbou či doloženo publikovanými vědeckými důkazy a to v některém ČS EU c)Dodatečné diagnostické a monitorovací postupy představují minim. riziko či zátěž pro bezpečnost SH v porovnání s běžnou klinickou praxí dotčeného ČS 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 21 Nové rozdělení studií – nové definice

22
Dokumentace KH – Part I a Part II PART I – posuzují všechny dotčené ČS společně Protokol, Farmaceutická dokumentace, Investigator´s Brochure PART II – posuzuje každý ČS samostatně; v průběhu posuzování spolu nekomunikují - způsob posouzení na ČS Informace pro pacienta / Informovaný souhlas Odměňování, kompenzace – zkoušejícím a SH Nábor SH Ochrana osobních údajů Zkoušející Místa klinického hodnocení Odškodnění / pojištění Souhlas s platnými pravidly pro shromažďování, skladování a budoucí použití biologických vzorků SH 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 22

23
Procedura povolení KH – Part I Jednotná žádost předložena přes portál Označeny dotčené ČS Zadavatel určí „Reporting member state“ (RMS) - ČS-zpravodaj Může vzít jiný ČS; Pokud nebude chtít žádný ČS, zůstane RMS ČS navržený zadavatelem CTAG (Koordinační a poradní skupina pro KH) – pravidla pro stanovování Reporting MS RMS – jako jediný pro Part I komunikuje se zadavatelem Součástí posouzení ČS – i etické → zapojení EK !!! Timeline pro povolení = 60 dnů 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 23

24
Procedura povolování KH – PART I - timeline 10 dnů validace Do 26 dne draft HZ 12 dnů připomínky ČS 7 dnů konsolidace 5 dnů 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 24 31 dnů = 12 zadavatel + 19 ČS (12 zhodnocení doplněné dokumentace + 7 konsolidace) Celkem 60 dnů - lze prodloužit o 50 dnů pro LPMT Spolupráce EK na posouzení Protokolu a IB (30 dnů?) Spolupráce EK na posouzení odpovědi (12 dnů) Finální HZ dotč. ČS zadavateli draft HZ dotč. ČS NEDODRŽENÍ ČASU: ZADAVATELEM → ŽÁDOST SE POVAŽUJE ZA STAŽENOU ČS → ŽÁDOST S POVAŽUJE ZA ÚPLNOU NEBO SCHVÁLENOU „TACIT APPROVAL“ Do 3.dne ČS zpravodaj Do 7 dnů informace ČSs REPORTING DATE
 Celkem 60 dnů - lze prodloužit o 50 dnů pro LPMT Spolupráce EK na posouzení Protokolu a IB (30 dnů ) Spolupráce EK na posouzení odpovědi (12 dnů) Finální HZ dotč. ČS zadavateli draft HZ dotč. ČS NEDODRŽENÍ ČASU: ZADAVATELEM → ŽÁDOST SE POVAŽUJE ZA STAŽENOU ČS → ŽÁDOST S POVAŽUJE ZA ÚPLNOU NEBO SCHVÁLENOU „TACIT APPROVAL Do 3.dne ČS zpravodaj Do 7 dnů informace ČSs REPORTING DATE.")
25
Procedura povolování KH – timeline PART II 10 dnů validace 45 dnů posouzení PART II 5 dnů 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 25 31 dnů prodloužení na doplnění 1x = 12 zadavatel odpověď + 19 ČS posouzení odpovědi Celkem 60 dnů - lze prodloužit o 50 dnů pro LPMT Společná validace (?) Jednotné stanovisko NEDODRŽENÍ ČASU - kdykoli: ZADAVATELEM → ŽÁDOST SE POVAŽUJE ZA STAŽENOU ČS → ŽÁDOST S POVAŽUJE ZA ÚPLNOU NEBO SCHVÁLENOU „TACIT APPROVAL“ Finální HZ 10 dnů na doplnění = 5 dnů zadavatel + 5 dnů ČS REPORTING DATE
 Jednotné stanovisko NEDODRŽENÍ ČASU - kdykoli: ZADAVATELEM → ŽÁDOST SE POVAŽUJE ZA STAŽENOU ČS → ŽÁDOST S POVAŽUJE ZA ÚPLNOU NEBO SCHVÁLENOU „TACIT APPROVAL Finální HZ 10 dnů na doplnění = 5 dnů zadavatel + 5 dnů ČS REPORTING DATE.")
26
Konečné vyjádření k žádosti o KH Povolení Povolení s podmínkou (pouze to, co nemohou v době povolení mít) Nesouhlas SH by obdržel horší léčbu než v běžné praxi Rozpor s národní legislativou Nesouhlas ČS s HZ RMS, co se týká bezpečnosti SH a spolehlivosti a robustnosti dat Nesouhlas – ČS sdělí zadavateli / EC / ostatním dotčeným ČS Nesouhlas RMS → platí pro všechny dotčené ČS Nesouhlas ČS– nesouhlas s rozhodnutím RMS nebo nesouhlas EK daného ČS → platí pro ČS ČS by měl nastavit možnost odvolání – v případě vydání nesouhlasu s KH !!! 5 dnů – finální stanovisko ČS (jednotné za ČS) 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 26 Návrh nařízení – hlavní obsah
 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 26 Návrh nařízení – hlavní obsah.")
27
Posouzení KH – PART I a PART II Nepředloží-li zadavatel současně – může Part II předložit – nejpozději do 2 let po schválení Part I (+ přiložit prohlášení, že Part I beze změn) Nezahájí-li zadavatel KH do 2 let od vydání povolení – povolení propadá; CTA by byla znovu posuzována – prošla by znovu celou procedurou Stažení žádosti zadavatelem – pouze do „reporting date“ Resubmition – opětovné podání po předchozím zamítnutí či stažení žádosti (žádost se považuje za novou) Přiřazení dalších ČS – (2.vlna posouzení) Vede stejný RMS 52 dnů - stejná procedura 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 27

28
Významné změny (dříve SA) Včetně změny centra a hl. zkoušejícího Part I Společně dotčenými ČS ; vede ČS-zpravodaj Assessment Report Part II Posuzuje ČS samostatně Současně Part I a Part II Kombinace výše uvedeného 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 28

29
Významná změna (VZ)- timeline PART I 6 dnů Validace VZ 19 dnů draft HZ stanovisko 12 dnů koordinace posouzení 3. 6. 2014 29 31 dnů prodloužení na doplnění 1x = 12 zadavatel odpověď + 19 ČS posouzení odpovědi Celkem 44 dnů NEDODRŽENÍ ČASU - kdykoli: ZADAVATELEM → ŽÁDOST SE POVAŽUJE ZA STAŽENOU ČS → ŽÁDOST S POVAŽUJE ZA ÚPLNOU NEBO SCHVÁLENOU „TACID APPROVAL“ 38.Den Finální HZ 10 dnů na doplnění 1x = 5 zadavatel doplní + 5 ČS posouzení doplnění Do 5.dne ČS připom. ČS-zpravodaji 38 dnů posouzení lze prodloužit o 50 dnů pro LPMT REPORTING DATE © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV

30
Významná změna (VZ)- timeline PART II 6 dnů Validace VZ 38 dnů posouzení VZ PART II stanovisko 3. 6. 2014 30 31 dnů prodloužení na doplnění 1x = 12 zadavatel odpověď + 19 ČS posouzení odpovědi Celkem 44 dnů NEDODRŽENÍ ČASU - kdykoli: ZADAVATELEM → ŽÁDOST SE POVAŽUJE ZA STAŽENOU ČS → ŽÁDOST S POVAŽUJE ZA ÚPLNOU NEBO SCHVÁLENOU „TACIT APPROVAL“ Finální HZ 10 dnů na doplnění 1x = 5 zadavatel doplní + 5 ČS posouzení doplnění REPORTING DATE © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV

31
Informovaný souhlas a ochrana SH Obecné požadavky platné pro všechny IP / IS Nejen písemný, nelze-li i video či audio záznam + podpis nezávislého svědka Pro nezletilé – dle národní legislativy Zvl. požadavky pro „zranitelné subjekty“ – samostatně Nezletilí Osoby zbavené právní způsobilosti Těhotné a kojící Naléhané situace (Emergency situation) Přípustné rozdíly dle národní legislativy Vojáci Vězni Osoby v ústavní péči Cluster study – požadavky zjednodušeného IS 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 31
 Přípustné rozdíly dle národní legislativy Vojáci Vězni Osoby v ústavní péči Cluster study – požadavky zjednodušeného IS © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 31.")
32
Průběh klinického hodnocení Průběh KH – co hlásí zadavatel Zahájení KH (1.akt náboru SH) Zařazení 1 SH v ČS Ukončení náboru SH v daném ČS Ukončení, předčasné ukončení Dočasné přerušení Pozastavení Bezpečnostní hlášení EudraVigilance EMA vytvoří webový formulář pro hlášení SUSAR do databáze NÚ Časové rozvržení hlášení Adverse Event Annual Reporting – posuzování všemi ČS – spolupráce 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 32

33
Další kapitoly Odškodnění ČS určí systém odškodnění – pojištění, náhrady škody Zadavatel a zkoušející musí dodržet ČS nesmí požadovat další odškodnění/pojištění, pokud je léčba v KH s malým rizikem a je kompenzována jinak Poplatky – stanoveny ČS, 1 za ČS Doporuč. redukovat pro nekomerční subjekty Požadavky na uchovávání dat / dokumentace (25 let od skončení KH) Požadavky na IMP a AMP, labeling Sledování, skladování, vracení a likvidace LP v KH 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 33
 Požadavky na IMP a AMP, labeling Sledování, skladování, vracení a likvidace LP v KH © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 33.")
34
Další kapitoly Urgent Safety Measure / naléhavá bezpečnostní opatření Inspekce velký důraz, dostatečné početní zajištění inspektorů EC – požadavky na kvalifikaci a tréning CTAG – Clinical Trials Coordination and Advisory Group Při Evropské komisi → předseda z EC Sekretariát bude zajišťovat EC Členy – zástupci jednotlivých ČS 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 34 Návrh nařízení – hlavní obsah

35
Nařízení pro KH Toto nařízení vstupuje v platnost 20. dnem po vyhlášení v Úředním věstníku Evropské unie. Toto nařízení se použije nejdříve 6 měsíců po zveřejnění oznámení podle čl. 82 odst. 3, ale v žádném případě ne dříve než 2 roky po zveřejnění tohoto nařízení. Toto nařízení je závazné v celém rozsahu a přímo použitelné ve všech členských státech. Přechodné období – KH předložená dle směrnice 2001/20/ES se nadále řídí tímto předpisem – 3 roky od vstupu v platnost nařízení V nařízení v čl. 98 bude uvedena i doba, kdy lze ještě předkládat žádosti dle směrnice a řídit se jejími požadavky. 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 35 Nařízení KH

36
Nejzásadnější změny v regulaci KH Jednotný portál – nová databáze KH Nastavení procesu posouzení v ČS – systém etických komisí Striktně dané časy – kalendářní dny – hrozba tichého souhlasu Nové dělení - klinické studie, KH a nízkointervenční KH RMS – hodnotící zprávy (AR) Posouzení dokumentace – změna systému Part I – společné všemi ČS Part II – národní, samostatně bez spolupráce Významné změny – hodnotící zprávy Změny v zajištění pojištění / odškodnění Poplatky Hodnotící zprávy pro Roční bezpečnostní zprávy
 Posouzení dokumentace – změna systému Part I – společné všemi ČS Part II – národní, samostatně bez spolupráce Významné změny – hodnotící zprávy Změny v zajištění pojištění / odškodnění Poplatky Hodnotící zprávy pro Roční bezpečnostní zprávy")
37
Dopad nařízení KH v ČR Legislativa Národní systém pojištění/odškodnění Systém etických komisí Proces posuzování KH Poplatky Správní řád Etické komise posuzující KH Technické zajištění činnosti Návaznost na portál EU Zajištění posuzování etické komise – zapojení / komunikace Personální Finanční Akademický výzkum

38
Počty předložených žádostí o KH Významné dodatky > 2000 / rok Počet předložených KH za rok 2014 ke dni 30. 5.2014
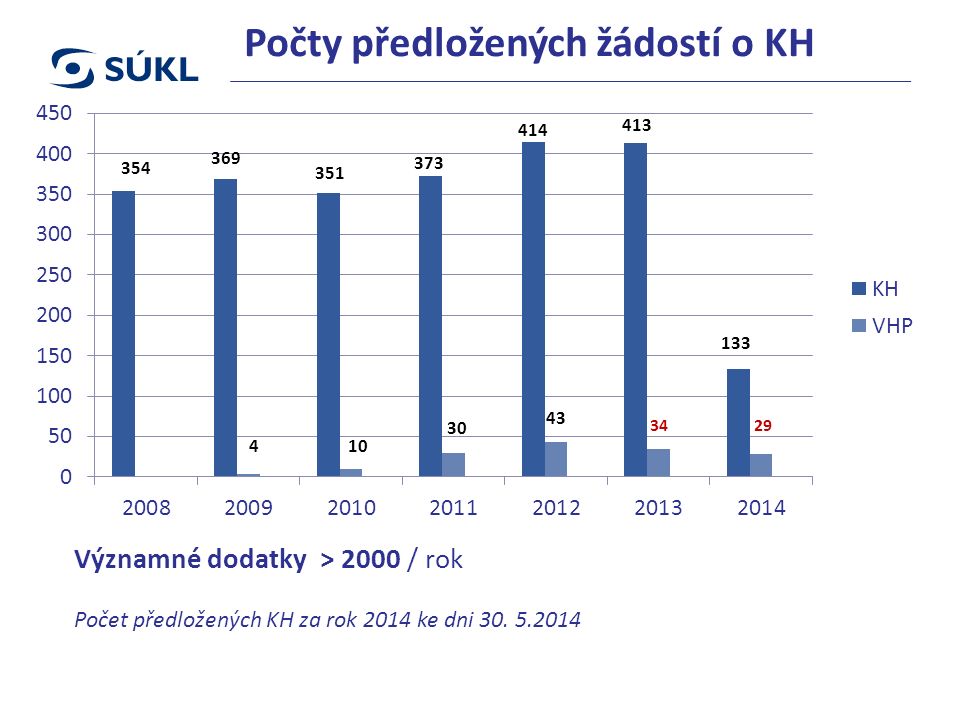
39
Děkuji za pozornost Dotazy - diskuse 3. 6. 2014 © 2012 STÁTNÍ ÚSTAV PRO KONTROLU LÉČIV 39

Podobné prezentace














 Zákon č. 100/2001 Sb., o posuzování vlivů na životní prostředí, ve znění zákona.>")




