Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Kapalinová chromatografie v analytické toxikologii Věra Pacáková Univerzita Karlova v Praze, Přírodovědecká fakulta, katedra analytické chemie

2
Obsah 1. Úvod 2. Principy HPLC 3. Instrumentace 4. Úprava vzorku 5. Analýza drog a jejich metabolitů 6. Závěry

3
1. Úvod GC – obvykle vyžaduje derivatizaci, bezvodé vzorky HPLC - doplňuje a nyní i nahrazuje GC Výhody – 85 % všech látek lze stanovit (tepelně labilních, polárních, nízko- i vysoko- molekulárních), přímá analýza bez derivatizace – časová úspora, přímý nástřik vodných vzorků Ve spojení s citlivou detekcí – MS – možnost identifikace a kvantifikace Nevýhody: pomalá difúze v kapalinách, nižší účinnost než GC, vyšší finanční náklady na analýzu (nákladnější přístroj, rozpouštědla)
, přímá analýza bez derivatizace – časová úspora, přímý nástřik vodných vzorků Ve spojení s citlivou detekcí – MS – možnost identifikace a kvantifikace Nevýhody: pomalá difúze v kapalinách, nižší účinnost než GC, vyšší finanční náklady na analýzu (nákladnější přístroj, rozpouštědla).")
4
2. Principy HPLC Mobilní fáze: kapalina složení mobilní fáze ovlivňuje separaci Stacionární fáze: chemicky vázané fáze adsorbenty měniče iontů gely (pro SEC) afinitní fáze Vysoká účinnost a rychlost analýzy – vysoké tlaky a malé částice s jednotnou velikostí, homogenně naplněné v koloně
 afinitní fáze Vysoká účinnost a rychlost analýzy – vysoké tlaky a malé částice s jednotnou velikostí, homogenně naplněné v koloně.")
5
Chromatogram (kvalita, kvantita, účinnost separace) t R – retenční čas, t´ R – redukovaný čas, plocha nebo výška píku, w – šířka píku
 t R – retenční čas, t´ R – redukovaný čas, plocha nebo výška píku, w – šířka píku")
6
Závislost H na lineární průtokové rychlosti mobilní fáze u H (výškový ekvivalent teoret. patra) = L/n n (počet pater) = 16(t R /w) 2
 = L/n n (počet pater) = 16(t R /w) 2.")
7
Závislost H na velikosti částic Čím účinnost separace 2 m 3 m 5 m 8 m

8
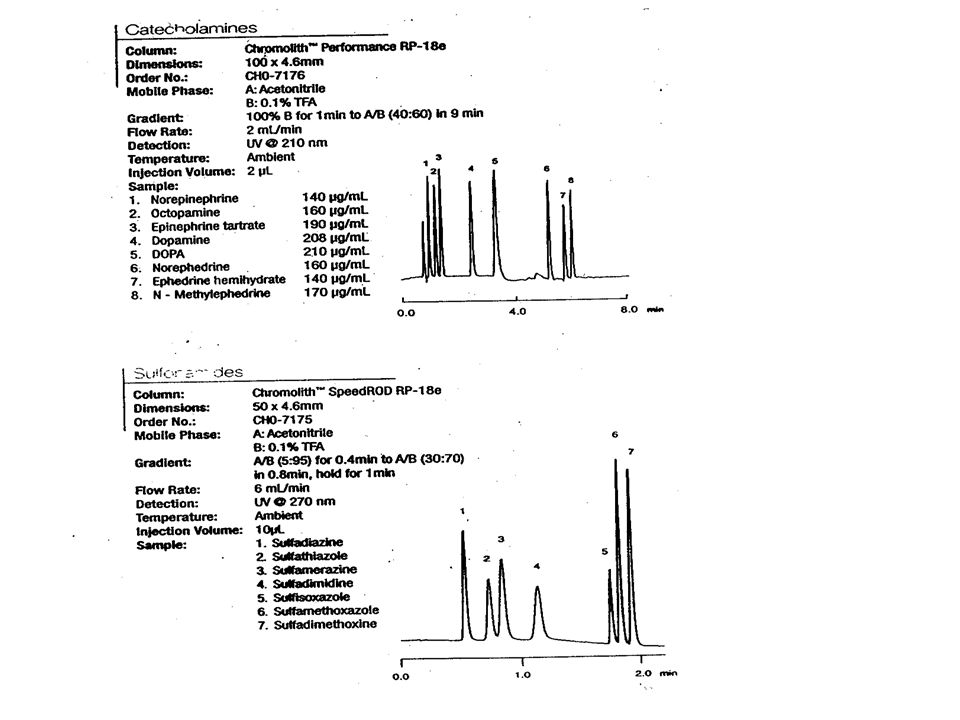
Volba kolony Náplně: zcela porézní částice 2- 10 m, velikost pórů 10 – 50 nm, pravidelného kulovitého tvaru, homogenně naplněné v koloně pelikulární – polopropustné (nepropustné jádro, tenká pórovitá vrstva nepórovité – rychlá sorpce, malá zátěž, póry 0,2 – 0,4 nm nedostupné pro solut monolitické kolony, vtištěné polymery Specifický povrch zátěž (dávkování) póry 10 nm 170 m 2 30 nm 100 m 2 nepórovité 0,6 – 6 m 2 /g
 póry 10 nm 170 m 2 30 nm 100 m 2 nepórovité 0,6 – 6 m 2 /g")
9
Miniaturizace ( HPLC) Zmenšování velikosti částic a průměru kolony Výhody: snížení spotřeby a odpadu rozpouštědel, stacionární fáze a vzorku vyšší účinnost separace vyšší citlivost detekce kompatibilita s MS detekcí Průměr kolonyPrůtok 4,5 mm1 mL/min 1 mm0,047 mL/min 0,25 mm0,003 mL/min
 Zmenšování velikosti částic a průměru kolony Výhody: snížení spotřeby a odpadu rozpouštědel, stacionární fáze a vzorku vyšší účinnost separace vyšší citlivost detekce kompatibilita s MS detekcí Průměr kolonyPrůtok 4,5 mm1 mL/min 1 mm0,047 mL/min 0,25 mm0,003 mL/min")
10
Chemicky vázané fáze Univerzální, vhodné pro biologické vzorky, rychlé ustavování rovnováhy- vysoká separační účinnost Nosič (silikagel, organický polymer) s navázanými funkčními skupinami: alkyly, zejména oktyl, oktadecyl, fenyl – nepolární (reverzní) fáze – nejvýznamnější (RPLC) normální fáze – polární – méně používané iontově výměnné skupiny -SO 3 H, - COOH, -NH 2, -N + (R) 3 chirální stacionární fáze (cyklodextriny a další)
 s navázanými funkčními skupinami: alkyly, zejména oktyl, oktadecyl, fenyl – nepolární (reverzní) fáze – nejvýznamnější (RPLC) normální fáze – polární – méně používané iontově výměnné skupiny -SO 3 H, - COOH, -NH 2, -N + (R) 3 chirální stacionární fáze (cyklodextriny a další)")
11
Struktura chemicky vázaných fází

12
Mobilní fáze Mobilní fáze není inertní, ovlivňuje separaci Možnosti změny složení mobilní fáze jsou prakticky neomezené Obecné požadavky: dobrá rozpustnost pro soluty, kompatibilita s detekcí (UV hrana), toxicita, viskosita, těkavost reverzní fáze – voda + organická rozpouštědla (pufry, pH, iontově párová činidla…) retence (log k) ~ obsah org. rozpouštědla nepolární fáze – nepolární organické rozpouštědlo + polární modifikátor (<1 %) měniče iontů – pufry Izokratická vs. gradientová eluce (obdoba programování teploty v GC)
 měniče iontů – pufry Izokratická vs. gradientová eluce (obdoba programování teploty v GC).")
13
Derivatizace Derivatizací měníme fyzikální a chemické vlastnosti analytů. Hlavní důvody pro převedení analytů na deriváty jsou tyto: zlepšení detekovatelnosti (nejčastěji v kombinaci s fluorescenčním detektorem) zvýšení těkavosti (v GC) zlepšení chromatografických vlastností (např. změna polarity), zlepšení stability analytů, umožnění chirální separace, změna matrice pro lepší separaci.
 zvýšení těkavosti (v GC) zlepšení chromatografických vlastností (např. změna polarity), zlepšení stability analytů, umožnění chirální separace, změna matrice pro lepší separaci..")
14
Kvalitativní analýza k = (t R – t M ) /t M (retenční poměr) r 12 = 12 = t´ R2 /t´ R1 = k 2 /k 1 (relativní retence, selektivita) Reprodukovatelnost: složení mobilní fáze, pracovní teplota, příprava stacionární fáze
 /t M (retenční poměr) r 12 = 12 = t´ R2 /t´ R1 = k 2 /k 1 (relativní retence, selektivita) Reprodukovatelnost: složení mobilní fáze, pracovní teplota, příprava stacionární fáze")
15
Kvantitativní analýza Zdroje chyb: Odběr reprezentativního vzorku Úprava vzorku (nejvýznamnější) Dávkování (1-2 %) Stacionární fáze Instrumentace, zpracování signálu a interpretace
 Dávkování (1-2 %) Stacionární fáze Instrumentace, zpracování signálu a interpretace")
16
Pracovní techniky při vyhodnocování chromatogramů Vnitřní normalizace: xi = (A i / A j ) 100 Absolutní kalibrace mi = (A i /A s ) m s Vnitřní standardizace mi = (RMR sr /RMR ir ) (A i /A s ) (V s / V i ) m s Metoda standardního přídavku A – plocha m – množství V i, V s – objemy při ředění v i,v s – dávkované objemy vzorku a stand. RMR – relativní mol. odezva

17
3. Instrumentace Blokové schéma chromatografu 1 - zdroj mobilní fáze, 2 – čerpadlo, 3 – dávkovač, 4 - kolona, 5 - detektor, 6,7 - zařízení pro zpracování signálu detektoru

18
Zásobník m.f. – uzavřená nádoba, odplynit čerpadla – bezpulzní, vysokotlaká, průtoky od l/min po mL/min dávkovače – smyčkové (vzorky v l) automatické dávkovače kolony – preparativní či analytické, náplňové i kapilární, vnitřní průměr od desítek m po jednotky mm, nerezové či skleněné detektory – spektrofotometrický (diode-array) refraktometrický, elektrochemický, fluorescenční, hmotnostní vyhodnocovací zařízení
 automatické dávkovače kolony – preparativní či analytické, náplňové i kapilární, vnitřní průměr od desítek m po jednotky mm, nerezové či skleněné detektory – spektrofotometrický (diode-array) refraktometrický, elektrochemický, fluorescenční, hmotnostní vyhodnocovací zařízení.")
19
Čerpadlo reciprokační

20
Schéma šesticestného dávkovacího ventilu se smyčkou a - plnění smyčky, b - vymývání smyčky do kolony 1, 4 - připojení smyčky, 2 - přívod mobilní fáze od čerpadla, 3 - připojení kolony, 5, 6 - odpad; nástřik v poloze 4

21
Kolony: náplňové 3 – 5 mm, ~ 1 mL/min mikronáplňové ~ 1 mm, ~ 20 - 60 L/min kapilární ~ 10 m, ~ nl/min Materiály: silikagel (pH 2-8, endcapping) alumina (pro bazické látky) organické polymery Monolitické kolony Vtištěné polymery
 alumina (pro bazické látky) organické polymery Monolitické kolony Vtištěné polymery")
22
Monolitické kolony až 97 % objemu kolony zaujímá stacionární fáze (oproti ca 70 % u náplňových kolon) vyšší účinnost separace velká porozita vysoký průtok, rychlé analýzy makropóry – 2 m velký průtok, malý tlakový spád mesopóry – 13 nm velký povrch pro sorpci analytů na bázi silikagelu nebo organických polymerů
 vyšší účinnost separace velká porozita vysoký průtok, rychlé analýzy makropóry – 2 m velký průtok, malý tlakový spád mesopóry – 13 nm velký povrch pro sorpci analytů na bázi silikagelu nebo organických polymerů")
24
Detektory UV/VIS – nejběžnější, s diodovým polem (DAD), téměř univerzální fluorescenční – citlivý, selektivní, derivatizace elektrochemický- citlivý i selektivní, omezené použití hmotnostní – nejvyšší vypovídací schopnost (kvalita i kvantita), vysoká citlivost a selektivita
, téměř univerzální fluorescenční – citlivý, selektivní, derivatizace elektrochemický- citlivý i selektivní, omezené použití hmotnostní – nejvyšší vypovídací schopnost (kvalita i kvantita), vysoká citlivost a selektivita")
25
UV/VIS detektor (a) schéma, (b) uspořádání mřížky
 schéma, (b) uspořádání mřížky")
26
Schéma fluorescenčního detektoru 1, 2 - vstup a výstup mobilní fáze, 3 - excitační záření, 4 - emitované záření, 5 ‑ vnější plášť cely, 6 - optický systém, 7 - cela (5 l), 8 - držák
, 8 - držák")
27
Blokové schéma hmotnostního spektrometru

28
Iontový zdroj - převedení analytu do ionizovaného stavu, fragmentace. Hmotnostní analyzátor rozděluje v prostoru nebo čase směs iontů o různých poměrech hmotnosti ku náboji (m/z), produkovanou v iontovém zdroji. Detektor poskytuje analogový signál úměrný počtu dopadajících iontů. Po digitalizaci převeden do počítače a zpracován do hmotnostních spekter. Hmotnostní spektrometr pracuje za velmi nízkých tlaků - výkonný vakuový čerpací systém. Vstup umožňující převedení vzorku do iontového zdroje. GC-MS nebo HPC-MS vhodné rozhraní (interface) snižující podíl mobilní fáze
, produkovanou v iontovém zdroji. Detektor poskytuje analogový signál úměrný počtu dopadajících iontů. Po digitalizaci převeden do počítače a zpracován do hmotnostních spekter. Hmotnostní spektrometr pracuje za velmi nízkých tlaků - výkonný vakuový čerpací systém. Vstup umožňující převedení vzorku do iontového zdroje. GC-MS nebo HPC-MS vhodné rozhraní (interface) snižující podíl mobilní fáze.")
29
Iontové zdroje Ionizační energie 7-16 eV Výtěžek ionizace kolem 0,01% Ionizační techniky měkké (nízká fragmentace) a tvrdé (vysoká fragmentace) GC Ionizace elektronem (electron ionization, EI) Chemická ionizace (chemical ionization, CI) HPLC Sprejové ionizační techniky v kapalné fázi, měkké termosprejová ionizace (thermospray, TS) elektrosprejová ionizace (electrospray, ES) chemické ionizace za atmosférického tlaku (atmospheric pressure chemical ionization, APCI )
 a tvrdé (vysoká fragmentace) GC Ionizace elektronem (electron ionization, EI) Chemická ionizace (chemical ionization, CI) HPLC Sprejové ionizační techniky v kapalné fázi, měkké termosprejová ionizace (thermospray, TS) elektrosprejová ionizace (electrospray, ES) chemické ionizace za atmosférického tlaku (atmospheric pressure chemical ionization, APCI )")
30
Schéma iontového zdroje ES

31
Schéma iontového zdroje APCI

32
Analyzátory Magnetický hmotnostní analyzátor Kvadrupólový analyzátor Iontová past (ion-trap) Průletový analyzátor (time of flight, TOF)
 Průletový analyzátor (time of flight, TOF)")
33
Interface Rozhraní s pohybujícím se kovovým páskem (moving belt interface) sprejové ionizace – většina těkavých látek odvedena mimo MS – bez interface mikroHPLC - přímý vstup MS/MS-LC systém se třemi kvadrupóly: První pro výběr mateřského iontu, do druhého se přivádí pod tlakem inertní plyn a slouží jako kolizní cela pro řízenou fragmentaci mateřského iontu, skenováním třetího kvadrupólu se rozdělí vzniklé dceřiné ionty
 sprejové ionizace – většina těkavých látek odvedena mimo MS – bez interface mikroHPLC - přímý vstup MS/MS-LC systém se třemi kvadrupóly: První pro výběr mateřského iontu, do druhého se přivádí pod tlakem inertní plyn a slouží jako kolizní cela pro řízenou fragmentaci mateřského iontu, skenováním třetího kvadrupólu se rozdělí vzniklé dceřiné ionty")
34
Rozhraní (interface) s pohybující se kovovou smyčkou
 s pohybující se kovovou smyčkou")
35
Hybridní tandemový hmotnostní spektrometr s dvojicí kvadrupólů a průletovým hmotnostním analyzátorem

36
Požadavky na HPLC-MS systém Kolona – malý průměr, malé průtokové rychlosti přímý převod do MS (bez interface) Mobilní fáze těkavá, omezit obsah solí, protože snižují výtěžek ionizace a zanášejí iontový zdroj Vhodnější methanol než acetonitril, vyšší výtěžek ionizace
 Mobilní fáze těkavá, omezit obsah solí, protože snižují výtěžek ionizace a zanášejí iontový zdroj Vhodnější methanol než acetonitril, vyšší výtěžek ionizace")
37
4. Úprava vzorku – přečištění, zakoncentrování Kapalné vzorky (moč, serum, sliny): extrakce kapalinou (LLE) extrakce tuhou fází (SPE) – nejběžnější materiály: chemicky vázané fáze, ionexy, SEC fáze, vtištěné polymery, atd. mikroextrakce na vláknech nebo v kapiláře (SPME) Tuhé vzorky (tkáně, vlasy): Soxhlet zrychlená extrakce rozpouštědly (ASE) extrakce ultrazvukem extrakce nadkritickými tekutinami (SFE)
: extrakce kapalinou (LLE) extrakce tuhou fází (SPE) – nejběžnější materiály: chemicky vázané fáze, ionexy, SEC fáze, vtištěné polymery, atd. mikroextrakce na vláknech nebo v kapiláře (SPME) Tuhé vzorky (tkáně, vlasy): Soxhlet zrychlená extrakce rozpouštědly (ASE) extrakce ultrazvukem extrakce nadkritickými tekutinami (SFE).")
38
SPME Solid phase micro- extraction

39
a-SPME extrakce, b-desorpce v GC, c-desorpce v HPLC

40
Přímé spojení SPME v kapiláře s HPLC a) extrakce
 extrakce")
41
b) Desorpce
 Desorpce")
42
Příprava vtištěných polymerů

43
5. Analýza drog a jejich metabolitů Analýza tělních tekutin Analýza krve - deproteinizace extrakce rozpouštědly (LLE) extrakce tuhou fází (SPE, SPME) přečištění extraktu na měničích iontů zakoncentrování odpařením vlastní analýza HPLC-ESI (API)-MS
 extrakce tuhou fází (SPE, SPME) přečištění extraktu na měničích iontů zakoncentrování odpařením vlastní analýza HPLC-ESI (API)-MS.")
44
Analýza vlasů Odběr vzorku – velká variabilita (místo odběru, stáří, pohlaví) - 10 – 250 mg vzorku dekontaminace vlasů (kosmetické přípravky, prach a pod.) extrakce, přečištění extraktu, zakoncentrování vlastní analýza
 - 10 – 250 mg vzorku dekontaminace vlasů (kosmetické přípravky, prach a pod.) extrakce, přečištění extraktu, zakoncentrování vlastní analýza")
45
(A) Chromatogram extraktu z krve oběti předávkované amobarbitalem (B) hmotnostní spektrum této látky
 Chromatogram extraktu z krve oběti předávkované amobarbitalem (B) hmotnostní spektrum této látky")
46
Rychlá HPLC-ESI-MS identifikace a kvantifikace významných nox Podmínky analýzy Gradientová eluce metanolem s 0,1% HCOOH Monolitická kolona Chromolith Detekce ESI-MS Doba analýzy 5 min. včetně stabilizace Všech 14 látek (neutrální, bazické, kyselé) jsou účinně ionizovány pozitivní ESI Detekční limity 10,0 až 50,0 ng/ml (K. Pihlainen et al., J. Chromatogr. A, 994 (2003) 93–102)
 jsou účinně ionizovány pozitivní ESI Detekční limity 10,0 až 50,0 ng/ml (K. Pihlainen et al., J. Chromatogr. A, 994 (2003) 93–102).")
47
( I) amfetamin, (II) 3,4-MDMA, (III) buprenorfin, (IV) clenbuterol, (V) salbutamol, (VI) LSD, (VII) metandienon, (VIII) nandrolon, (IX) stanozolol, (X) testosteron, (XI) morfin, (XII) f enobarbital, (XIII) psilocybin, (XIV) temazepam
 amfetamin, (II) 3,4-MDMA, (III) buprenorfin, (IV) clenbuterol, (V) salbutamol, (VI) LSD, (VII) metandienon, (VIII) nandrolon, (IX) stanozolol, (X) testosteron, (XI) morfin, (XII) f enobarbital, (XIII) psilocybin, (XIV) temazepam")
48
MS–MS spektra amfetaminu a 3,4-MDMA

49
A p-OH-AP, B p-OH-MA, C norefedrin D, efedrin, E AP; F 3,4- methylendioxyamfetamin (MDA), G 3,4-methylendioxymethamfetamin (MDMA), H MA, I methoxyfenamin, J DMA. K dibenzylamin (I.S.)
.")
50
(1) psilocybin (2) morfin (3) salbutamol (4) amfetamin (5) 3,4-MDMA (6) clenbuterol (7) LSD (8) fenobarbital (9) buprenorfin (10) temazepam (11) nandrolon, (12) metandienon (13) testosteron (14) stanozolol Rekonstruovaný iontový chromatogram
 psilocybin (2) morfin (3) salbutamol (4) amfetamin (5) 3,4-MDMA (6) clenbuterol (7) LSD (8) fenobarbital (9) buprenorfin (10) temazepam (11) nandrolon, (12) metandienon (13) testosteron (14) stanozolol Rekonstruovaný iontový chromatogram")
51
Analýza amfetaminů Mikroextrakce monolitickou kapilárou on-line s HPLC (UV detekce) pro analýzu amfetaminu, methamfetaminu a jejich methylendioxy- derivátů v moči D.L. 1.4–4.0 ng/mL. Vysoká reprodukovatelnost (RSD < 2.9%) v rozsahu 0.05–5 g/mL, doba analýzy 25 min. (Yi F. et al., J.Chromatogr.A, 1074 (2005) 9–16.)
 v rozsahu 0.05–5 g/mL, doba analýzy 25 min. (Yi F. et al., J.Chromatogr.A, 1074 (2005) 9–16.).")
52
Amfetamin (PA), methamfetamin (MPA), 3,4-methylendioxo- amfetamin (MDA), 3,4-methylendioxomethamfetamin (MDMA)
, methamfetamin (MPA), 3,4-methylendioxo- amfetamin (MDA), 3,4-methylendioxomethamfetamin (MDMA)")
53
Analýza vzorku s PA,MDA,MPA, MDMA (1 g/mL) s SPME (a) a přímý nástřik (b)
 s SPME (a) a přímý nástřik (b)")
54
Analýza moči 3 osob podezřelých ze závislosti na amfetaminech

55
Extrahovaný chromatogram vzorku moči pacienta zneužívajícího DMA 4 – MA 5 – AP 6 – DMA-N-oxid 7 –DMA

56
Extrahovaný chromatogram vzorku moči pacienta zneužívajícího selegilin 1 - SG-N-oxid 2 – desmethylselegilin 3 – ethylamfetamin 4 – MA 5 - AP

57
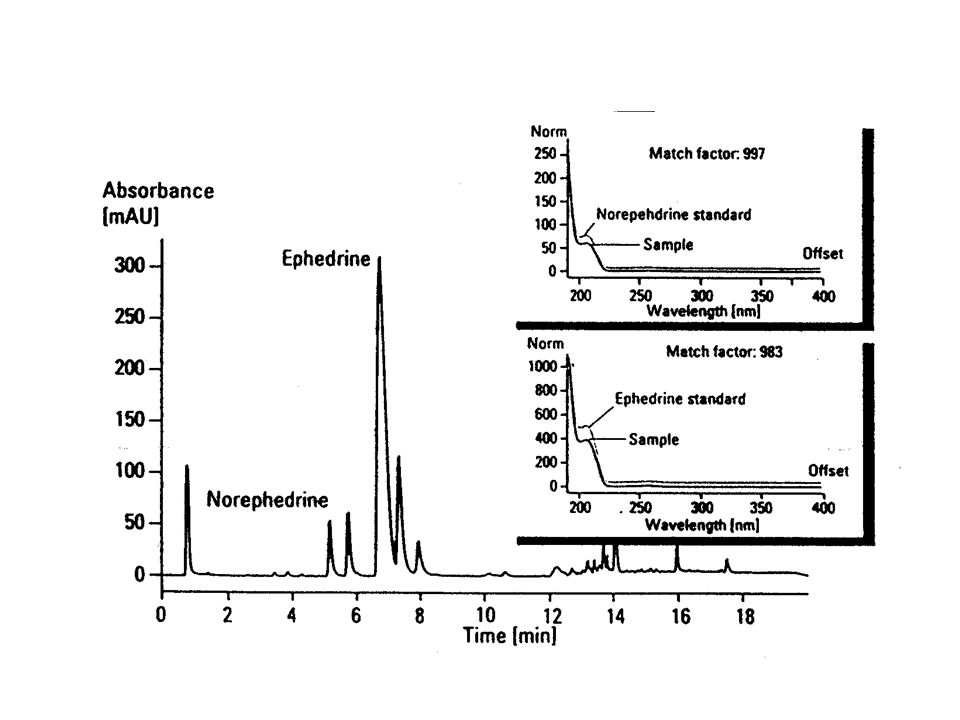
Struktura Fen, Norf a N-Fen N-Fen v čínských přípravcích k hubnutí – Fen a Norf- metabolity, škodlivé účinky, zakázané

58
A – extrakt vlasů, B – extrakt vlasů se 100 pg/mg norfenfluraminu a 230 pg/mg fenfluraminu, C - extrakt 1 vlasu pacienta (HPLC s fluorescenční detekcí po derivatizaci)
")
59
Stanovení meprobamatu ve vlasech Předběžná úprava: 1 M HCl přes noc při 60 o C Extrakce: LL s fosfátovým pufrem (pH 5,5) a chloroformem Analytická metoda: HPLC-MS nebo GC-MS pro derivatizaci s trimethylfenylamonium hydroxide Měřené koncentrace 3,32 and 4,21 ng/mg (2 osoby) DL 0,2 ng/mg [28] Dávka: 400 mg 4,27–6,08 ng/mg. 800 mg 8,54–13,01 ng/mg 1200 mg 11,89–17,64 ng/mg
![Stanovení meprobamatu ve vlasech Předběžná úprava: 1 M HCl přes noc při 60 o C Extrakce: LL s fosfátovým pufrem (pH 5,5) a chloroformem Analytická metoda: HPLC-MS nebo GC-MS pro derivatizaci s trimethylfenylamonium hydroxide Měřené koncentrace 3,32 and 4,21 ng/mg (2 osoby) DL 0,2 ng/mg [28] Dávka: 400 mg 4,27–6,08 ng/mg.](http://images.slideplayer.cz/9/2523025/slides/slide_59.jpg "800 mg 8,54–13,01 ng/mg 1200 mg 11,89–17,64 ng/mg.")
60
Sulfonylmočovinové antidiabetikum glibenclamid

61
Extrahovaný iontový chromatogram m/z 221, celkový iontový chromatogram a hmotnostní spektrum ethylglukuronidu (metabolit ethanolu)
")
62
Tetrodoxin - toxin z ryby ježíka

63
Stanovení halucinogeních aminů Psilocybin (4-fosforyloxy-N-dimethyltryptamin) Psilocin (4-hydroxy-N,N-dimethyltryptamin) HPLC s elektrochemickou detekcí, + 1,0 V (Ag/AgCl), mobilní fáze 0,1 M fosfátový pufr, pH 3,8 + 10 v/v ethanolu Obsah v houbě Psilocybe bohemica Šebek Psilocybin0,57 % Psilocin 0,061 %
 Psilocin (4-hydroxy-N,N-dimethyltryptamin) HPLC s elektrochemickou detekcí, + 1,0 V (Ag/AgCl), mobilní fáze 0,1 M fosfátový pufr, pH 3, v/v ethanolu Obsah v houbě Psilocybe bohemica Šebek Psilocybin0,57 % Psilocin 0,061 %")
64
HPLC analýza extraktu houby Psilocybe bohemica Šebek s UV a elektrochemickou detekcí (R.Kysilka a spol., J.Chromatogr. 320, 414 (1985))
).")
65
Antiepileptika – HPLC-MS (API-ES positive) kafein, fenylethylmalonamid,ethosuximid, primidon, fenobarbital, methylfenylsukcimid, karbamazepinepoxid, fenoytoin,karbamazepin )
 kafein, fenylethylmalonamid,ethosuximid, primidon, fenobarbital, methylfenylsukcimid, karbamazepinepoxid, fenoytoin,karbamazepin )")
66
HPLC-UV analýza Ginko biloby (SPE –C18, gradientová eluce methanol-H 3 PO 4 -voda

67
HPLC-DAD analýza extraktu rostliny Ephedra Sinica Stamp ( extrakce etherem, gradientová eluce acetonitril, H 3 PO 4,voda)
")
69
Chirální separace Důležité ve farmaceutickém průmyslu, ale i při stanvoení metabolitů Derivatizace s chirálním činidlem v kombinaci s nechirální stacionární fází Chirální stacionární fáze – chemicky vázaný cyklodextrin, ergotalkaloidy, albumin atd.

70
Extase (ADAM, E, XTC) MDMA – 3,4-methylendioxymethamfetamin 1912 – vyvinuto firmou Merck jako anoretikum, neuvedeno na trh euforické a stimulující účinky enantimery, S + forma je potentnější než R - forma chirální separace s chemicky vázanou fází (cyklodextrin)
 MDMA – 3,4-methylendioxymethamfetamin 1912 – vyvinuto firmou Merck jako anoretikum, neuvedeno na trh euforické a stimulující účinky enantimery, S + forma je potentnější než R - forma chirální separace s chemicky vázanou fází (cyklodextrin)")
71
Extrahovaný chromatogram (A) moči a MS spektra L-AP (B) a L-MA (C) 1, D-AP; 2, L-AP; 3, D-MA, 4 L-MA
 moči a MS spektra L-AP (B) a L-MA (C) 1, D-AP; 2, L-AP; 3, D-MA, 4 L-MA")
72
Závěry HPLC-MS stává rutinní metodou v analytické toxikologie Umožňuje identifikaci a stanovení drog a jejich metabolitů ve stopových koncentrací ve složité matrici Miniaturizace usnadňuje spojení HLPC-MS Nové stacionární fáze: zrychlení analýzy, vyšší selektivita

73
Doporučená literatura V. Pacáková, K. Štulík, Vysokoúčinná kapalinová chromatografie, SNTL, Praha 1986. K.Štulík a kol., Analytické separační metody, Karolinum, Praha 2004

Podobné prezentace