Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
DATABÁZE A VYHLEDÁVÁNÍ SEKVENCÍ
MARIAN NOVOTNÝ MOLEKULÁRNÍ TAXONOMIE 2010

2
PŘEDNÁŠEJÍCÍ Mgr. Marian NOVOTNÝ, PhD.
vystudoval odbornou biologii na PřF UK, diplomka v laboratoři doc. Folka doktorát na Uppsalské univerzitě se specializací strukturní bioinformatika (Gerard Kleywegt) Marie Curie Fellow na Evropském Bioinformatickém Institutu (Janet Thornton & Roman Laskowski) ornitolog amatér
 Marie Curie Fellow na Evropském Bioinformatickém Institutu (Janet Thornton & Roman Laskowski) ornitolog amatér.")
3
OSNOVA co je substrát pro molekulární taxonomii?
kde se shromažďují data? jak data vyhledávat ?

4
REKONSTRUKCE EVOLUČNÍ HISTORIE
rekonstrukce na základě srovnávání znaků v molekulární taxonomii se používají sekvence sekvence (DNA, RNA, proteiny) se srovnávají tzv. alignmentem
 se srovnávají tzv. alignmentem.")
5
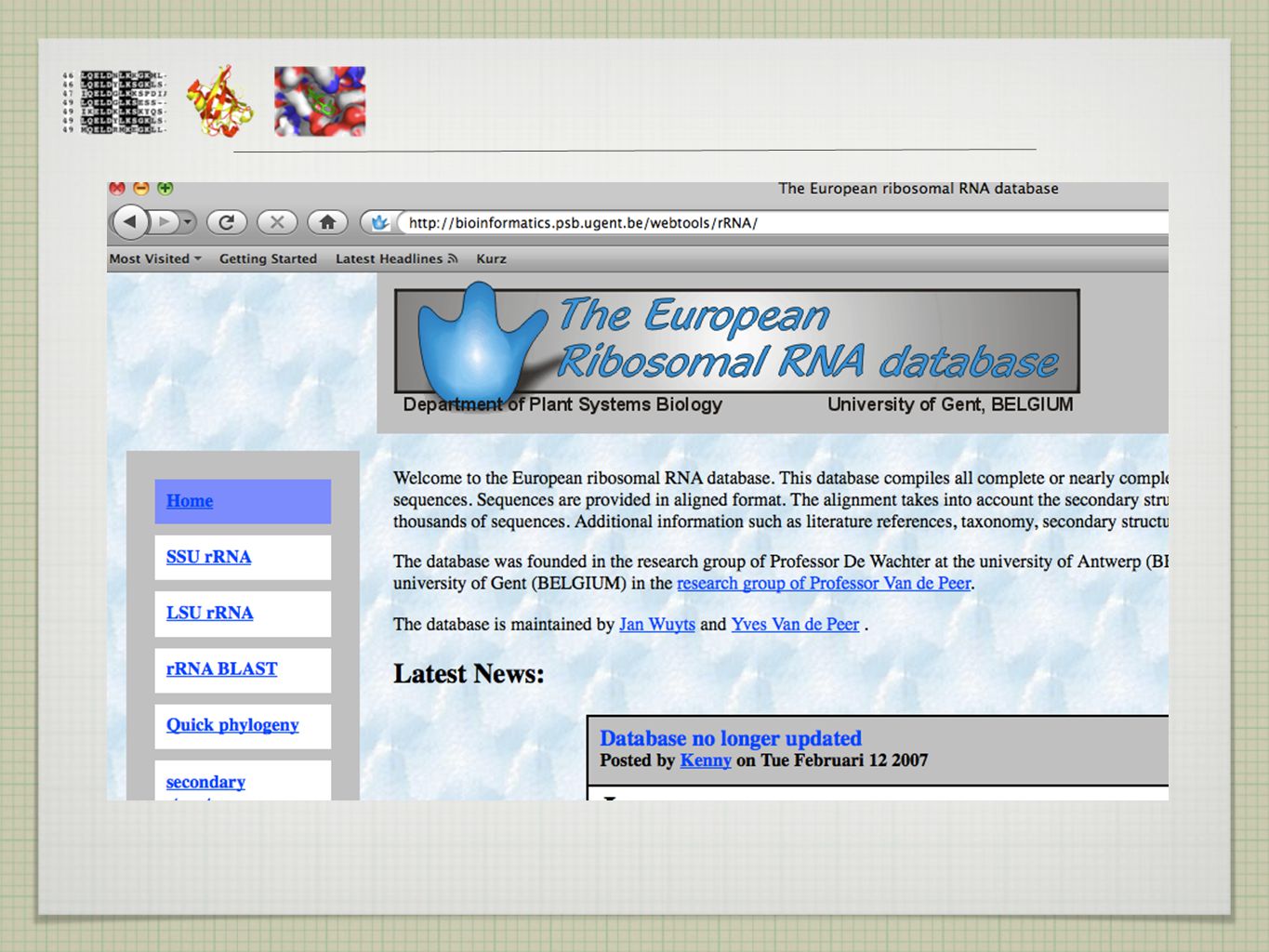
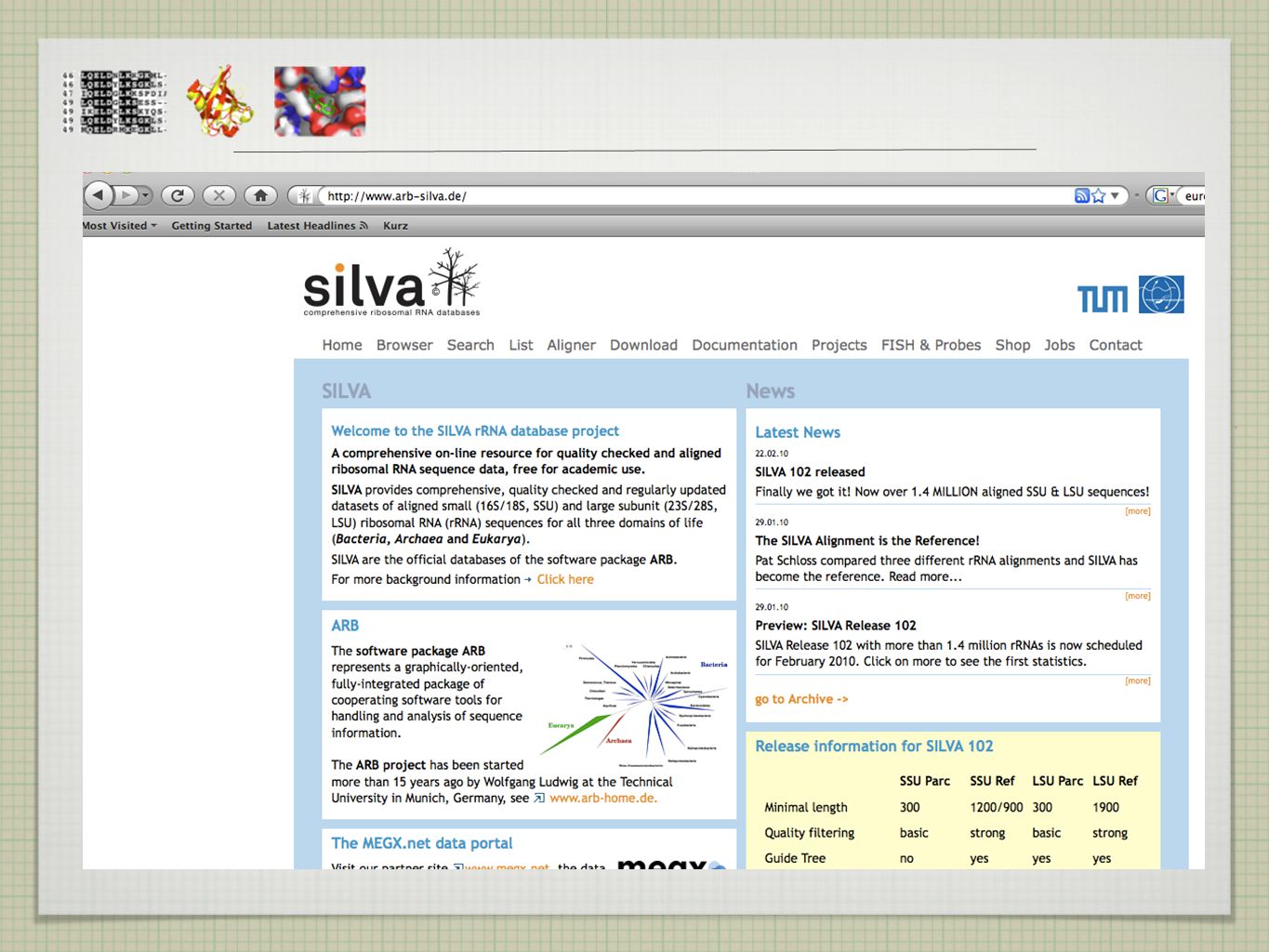
KDE NAJÍT SEKVENCE?

6
BIOINFORMATICKÉ DATABÁZE
úložiště dat (volně) dostupné pro kohokoliv snadno k nalezení lednové číslo Nucleid Acid Research (NAR)
 dostupné pro kohokoliv. snadno k nalezení. lednové číslo Nucleid Acid Research (NAR)")
7
LEDNOVÉ ČÍSLO NAR Nucleotide Sequence Databases RNA sequence databases
Protein sequence databases Structure Databases Genomics Databases (non-vertebrate) Metabolic and Signaling Pathways Human and other Vertebrate Genomes Human Genes and Diseases Microarray Data and other Gene Expression Databases Proteomics Resources Other Molecular Biology Databases Organelle databases Plant databases Immunological databases
 Metabolic and Signaling Pathways. Human and other Vertebrate Genomes. Human Genes and Diseases. Microarray Data and other Gene Expression Databases. Proteomics Resources. Other Molecular Biology Databases. Organelle databases. Plant databases. Immunological databases.")
8
VLASTNOSTI DATABÁZE četnost aktualizace dat
četnost aktualizace software redundance anotace dat anotace databáze

12
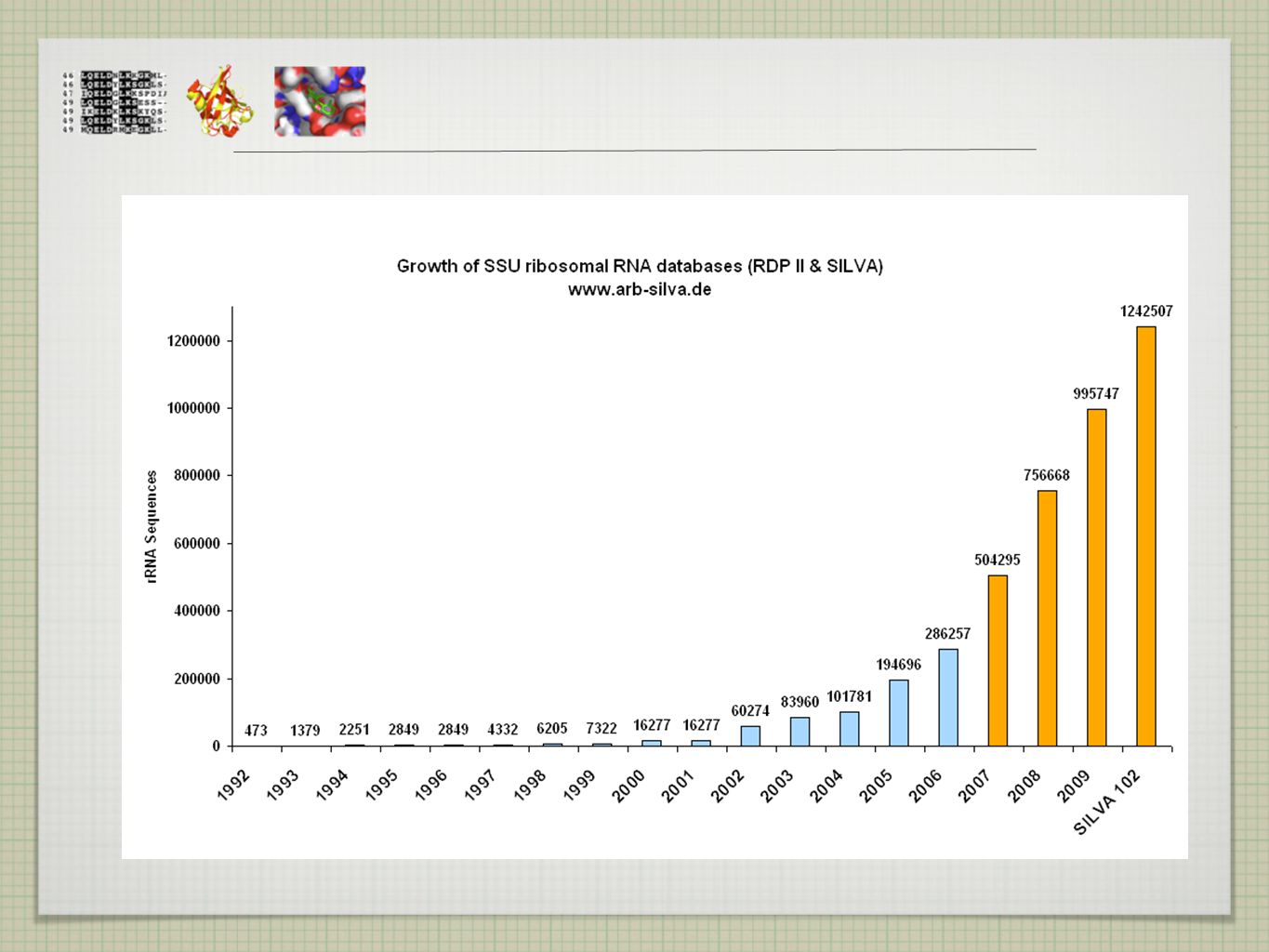
DNA DATABÁZE GenBank(NCBI) -112 Gb ve 112 mil. sekvencí - anotovaných
EMBL (EBI) Gb DDJB (Japonsko)
 Gb. DDJB (Japonsko)")
13
MÁLO ANOTOVANÁ SEKVENCE

14
ANOTOVANÁ SEKVENCE

16
GENOMOVÉ DATABÁZE http://www.ensembl.org/index.html

19
PROTEINOVÉ DATABÁZE Uniprot - Swissprot + TrEMBL - 10,5 mil. sek.
Swiss-prot - anotováno, ~ sekvencí GenPept - překládaný GenBank

20
UNIPROT + TREMBL AMINO ACID COMPOSITION
2.1 COMPOSITION IN PERCENT FOR THE COMPLETE DATABASE ALA (A) GLN (Q) LEU (L) SER (S) 6.72 ARG (R) GLU (E) LYS (K) THR (T) 5.61 ASN (N) GLY (G) MET (M) TRP (W) 1.31 ASP (D) HIS (H) PHE (F) TYR (Y) 3.06 CYS (C) ILE (I) PRO (P) VAL (V) 6.71 ASX (B) GLX (Z) XAA (X) 0.06
 8.57 GLN (Q) 3.88 LEU (L) 9.81 SER (S) ARG (R) 5.47 GLU (E) 6.14 LYS (K) 5.30 THR (T) ASN (N) 4.17 GLY (G) 7.08 MET (M) 2.45 TRP (W) ASP (D) 5.28 HIS (H) 2.20 PHE (F) 4.03 TYR (Y) CYS (C) 1.29 ILE (I) 6.00 PRO (P) 4.74 VAL (V) ASX (B) GLX (Z) XAA (X)")
22
JAK DATA VYHLEDÁVAT ?

23
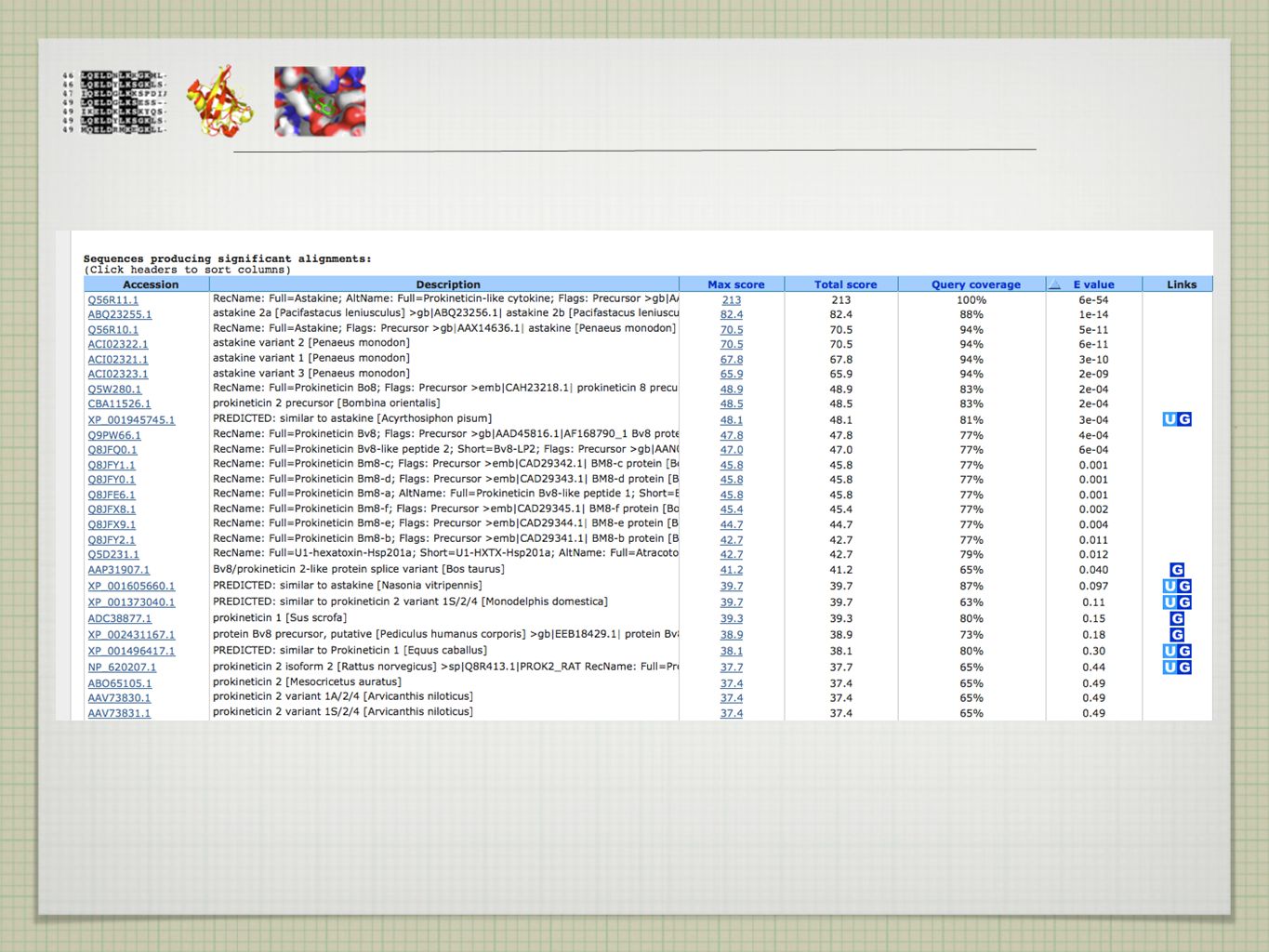
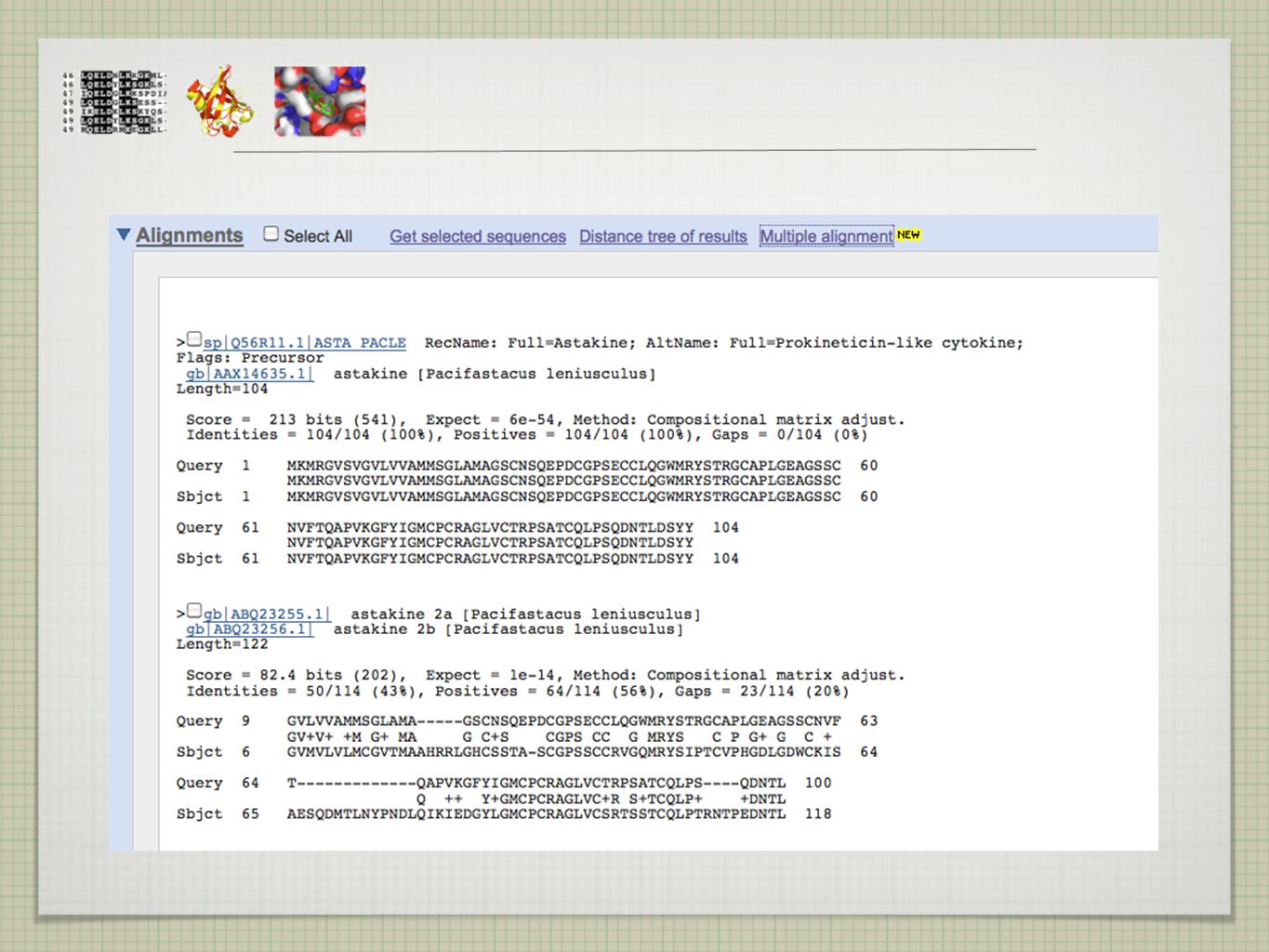
EXISTUJÍ PŘÍBUZNÉ SEKVENCE A KDE JE NAJÍT?
>ASTAKINE MKMRGVSVGVLVVAMMSGLAMAGSCNSQEPDCGPSECCLQGWMR YSTRGCAPLGEAGSSCNVFTQAPVKGFYIGMCPCRAGLVCTRPSATCQLPSQDNTLDSYY EXISTUJÍ PŘÍBUZNÉ SEKVENCE A KDE JE NAJÍT?

24
HLEDÁNÍ V DATABÁZÍCH - ALGORITMY
tradiční algoritmy (Needleman-Wunsch, Smith-Waterman) pomalé pro prohledávání velkých databází používány heuristické metody - rychle vede k výsledku, který se blíží optimálnímu řešení (ale nezaručuje jej) -> pro vyšší rychlost je obětována přesnost (rule of thumb) v případě sekvenčního srovnávání se metoda vzdává jistoty nalezení optimálního alignmentu, aby v krátkém čase provedla srovnání se všemi sekvencemi v databázi ( x rychlejší) klasickými heuristickými metodami jsou FASTA a BLAST obě metody použitelné pro DNA i proteinové sekvence
 pomalé pro prohledávání velkých databází. používány heuristické metody - rychle vede k výsledku, který se blíží optimálnímu řešení (ale nezaručuje jej) -> pro vyšší rychlost je obětována přesnost (rule of thumb) v případě sekvenčního srovnávání se metoda vzdává jistoty nalezení optimálního alignmentu, aby v krátkém čase provedla srovnání se všemi sekvencemi v databázi ( x rychlejší) klasickými heuristickými metodami jsou FASTA a BLAST. obě metody použitelné pro DNA i proteinové sekvence.")
25
FASTA metoda popsaná v 80. letech 20. století (Lipman & Pearson)
rychlá, heuristická metoda (na úkor senzitivity), globální alignment zjednodušení v první fázi, sekvence rozděleny na krátké úseky program generuje všechny možné “k-tuples” o délce k z dané sekvence k = 1-2 pro proteiny, k = 4-6 pro DNA k-tuples jsou porovnávány s k-tuples sekvencí v databázích
, globální alignment. zjednodušení v první fázi, sekvence rozděleny na krátké úseky. program generuje všechny možné k-tuples o délce k z dané sekvence. k = 1-2 pro proteiny, k = 4-6 pro DNA. k-tuples jsou porovnávány s k-tuples sekvencí v databázích.")
26
FASTA hledání SHOD v k-tuples
skórováni shod pomocí skórovací tabulky (Blosum 50) a rozšíření alignmentu (bez mezer) vysoce skórující shody vybrány vybere úseky, které budou součástí alignmentu dynamické programování pro konečný alignment (mezery)
 a rozšíření alignmentu (bez mezer) vysoce skórující shody vybrány. vybere úseky, které budou součástí alignmentu. dynamické programování pro konečný alignment (mezery)")
27
BLAST BLAST = Basic Local Alignment Search Tool Altschul et al., 1990
sekvence rozděleny na slova (words) a slova skórována vůči databázi všech slov slova skórována skórovací tabulkou (Blosum 62) a jen ty, které dosáhnou předem nadefinovaného minimálního skóre (treshold) jsou dále používány slova se skóre větším než treshold nemusí nutně obsahovat jen shody ( na rozdíl od Fasty) v prvním kroku se porovnávají slova bez mezer
 a slova skórována vůči databázi všech slov. slova skórována skórovací tabulkou (Blosum 62) a jen ty, které dosáhnou předem nadefinovaného minimálního skóre (treshold) jsou dále používány. slova se skóre větším než treshold nemusí nutně obsahovat jen shody ( na rozdíl od Fasty) v prvním kroku se porovnávají slova bez mezer.")
28
BLAST - HSP HSP - high scoring pair
vyber jen taková “slova”, která dosahují alespoň skóre X (treshold) PEG versus PQA PEQ má s Blosum 62 skóre 15, PQA jen 12 pokud si stanovíme treshold 13, tak budeme dále hledat jen slovo PEQ
 PEG versus PQA. PEQ má s Blosum 62 skóre 15, PQA jen 12. pokud si stanovíme treshold 13, tak budeme dále hledat jen slovo PEQ.")
29
BLAST II takto vybráná slova jsou hledána v databázi modifikovaným Smith- Watermanem (50 x rychlejší) HSP jsou dále rozšiřovány na obě strany dokud skóre roste v posledním kroku jsou nejlépe skórující páry (HSP`s) podrobeny dynamickému programování, které produkuje výsledné skóre a alignment vzhledem k rostoucí velikosti databází je třeba algoritmus neustále modifikovat (dvě shody v okně definované velikosti) obvykle citlivější než FASTA implementován jako server na řadě míst (NCBI, EBI)
 podrobeny dynamickému programování, které produkuje výsledné skóre a alignment. vzhledem k rostoucí velikosti databází je třeba algoritmus neustále modifikovat (dvě shody v okně definované velikosti) obvykle citlivější než FASTA. implementován jako server na řadě míst (NCBI, EBI)")
30
VERZE BLASTU blastn - hledá s DNA sekvencí (query) v DNA databázi
blastp - hledá s proteinovou sekvencí v proteinové databázi blastx - hledá s DNA sekvencí (6 rámců) v proteinové databázi tblastn - hledá s proteinovou sekvencí v DNA databázi tblastx - překládaná DNA v překládané DNA databázi megablast - víc query najednou
 v proteinové databázi. tblastn - hledá s proteinovou sekvencí v DNA databázi. tblastx - překládaná DNA v překládané DNA databázi. megablast - víc query najednou.")
33
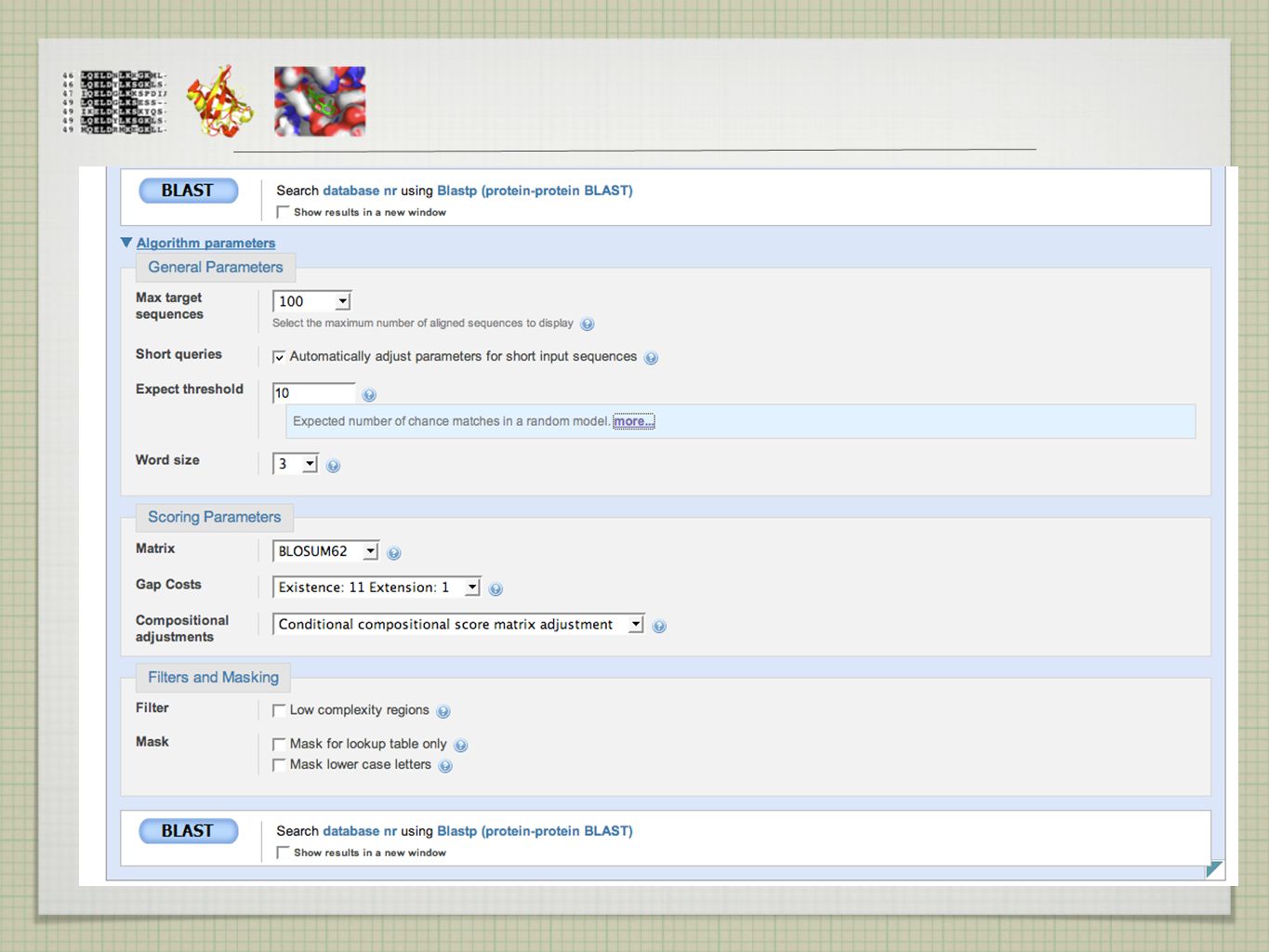
BLAST - VÝBĚR databáze - DNA x protein, anotovaná x kompletní, strukturní, genomové, specializované (protilátky) ... organismus datum - sekvence za poslední dva týdny skórovací tabulka - blosum 62 velikost slova low-complexity region filter - často P, D, N, E - false positive “default”nastavení algoritmu vhodné ve většině případů

34
BLOSUM BLOSUM 80 - tabulka vypočítaná na základě alignmentu bloku sekvencí s identitou 80 % BLOSUM 62 - tabulka vypočítaná na základě alignmentu bloku sekvencí s identitou 62 %

35
BLOSUM VERSUS PAM PAM 250 PAM 10 BLOSUM 90 BLOSUM 62 VELMI PŘÍBUZNÍ VZDÁLENĚ PŘÍBUZNÍ

36
VÝZNAMNOST NÁLEZU optimální alignment lze nalézt pro jakékoliv dvě sekvence dvě náhodné DNA sekvence = ~ 25% SI dvě náhodné proteinové sekvence = ~ 5% SI jak určit, že je alignment statisticky významný ?

37
PARAMETRY VÝZNAMNOSTI
P-value E-value pouze statistická významnost skóre -> biologickou relevanci záhodno ověřovat experimentálně

38
P-VALUE P-value - pravděpodobnost, že sekvence budou srovnány s nalezeným nebo vyšším skóre a zároveň nebudou příbuzné (false positive hit) P-value - pravděpodobnost, že bude skóre x nebo vyššího dosaženo náhodou pro účely výpočtu lze náhodu simulovat přeskládáváním sekvencí nebo výběrem vzorku z databáze druhá možnost lépe odpovídá realitě a poskytuje lepší výsledky (především u DNA)
")
39
EVD rozložení skóre lokálních alignmentů nepříbuzných sekvencí neodpovídá normálnímu rozdělení, ale rozdělení podle extrémních hodnot (EVD) při normálním rozdělení by docházelo k přeceňování významu dosažených skóre Dundas et al. BMC Bioinformatics 2007

40
P-VALUE P-value (S>x) = 1-exp (-exp (-λ(x-u))),
u = charakteristická hodnota = Kmn/λ m,n = délky sekvencí; K = konstanta; λ = “decay factor” K a λ mohou být kalkulovány z vlastností skórovací tabulky

41
E-VALUE E-value = pravděpodobnost, že bude dosaženo skóre x nebo vyššího náhodou v databázi dané velikosti E-value = P-value x N ; velikost databáze příklad: databáze o miliónu sekvencí a P-value = 10-6 cutoff (expect treshold) parametr v BLASTU - udává kolik lze průměrně očekávat false positives v databázi dané velikosti -> způsob jak vyvažovat senzitivitu a selektivitu nižší hodnota cutoff zvyšuje selektivitu, ale snižuje senzitivitu E-value = 10-6 x 106 =1
 parametr v BLASTU - udává kolik lze průměrně očekávat false positives v databázi dané velikosti -> způsob jak vyvažovat senzitivitu a selektivitu. nižší hodnota cutoff zvyšuje selektivitu, ale snižuje senzitivitu. E-value = 10-6 x 106 =1.")
42
BLAST / EVOLUČNÍ VZDÁLENOST
říká nám BLAST něco o příbuznosti nalezených sekvencí ? Je první “hit” evolučně nejpříbuznější query (hledané sekvenci)? BLAST většinou nalezá příbuzné sekvence nejpříbuznější sekvence však mohou chybět v databázi lokální alignment - často skóruje nejlépe vzdálené příbuzné 7 % sekvencí E.coli mělo nejlépe skórující sekvenci mimo Bacteria
 BLAST většinou nalezá příbuzné sekvence. nejpříbuznější sekvence však mohou chybět v databázi. lokální alignment - často skóruje nejlépe vzdálené příbuzné. 7 % sekvencí E.coli mělo nejlépe skórující sekvenci mimo Bacteria.")
45
2JTK

46
SEQUENCE IDENTITA/HOMOLOGIE
NEHOMOLOGNÍ PROTEINY ROST, 1999

47
SEQUENCE IDENTITA/HOMOLOGIE
HOMOLOGNÍ PROTEINY ROST, 1999

48
SEQUENCE IDENTITA/HOMOLOGIE
sekvenční identita > 35% - pravděpodobně homolog sekvenční identita = 20-35% (“twilight zone”; Doolittle) - může být homolog sekvenční identita < 20% - “midnight zone” (Rost) - sekvence zcela nedostatečná k určení homologie
 - může být homolog. sekvenční identita < 20% - midnight zone (Rost) - sekvence zcela nedostatečná k určení homologie.")
49
Average sequence identity of random alignments - 5.6 %
Sander et al., preprint Average sequence identity of random alignments % Average sequence identity of remote homologues %

50
SSEARCH pokud máte moře času nebo počítačový klastr nebo jste zoufalí
rigorózní Smith-Waterman - local alignment v databázi

51
DALŠÍ METODY HLEDÁNÍ V DATABÁZÍCH
profilové metody HMM modely

52
PROFILY modifikují skórovací tabulky specificky pro skupiny proteinů a pozici v alignmentu (např. globiny) pro každou pozici v alignmentu jsou generovány specifická skóre jak pro záměnu za jakoukoliv aa, tak pro inzerci nebo deleci Prof (pos,aa) = Σtype N(pos,type) x S(type, aa) x 10 N(pos,type) = podíl výskytu aa x na pozici y S(type, aa) = skóre skórovací tabulky pro zaměňovaný pár
 = Σtype N(pos,type) x S(type, aa) x 10. N(pos,type) = podíl výskytu aa x na pozici y. S(type, aa) = skóre skórovací tabulky pro zaměňovaný pár.")
53
PŘÍKLAD PROFILU v alignmentu globinů se na pozici 3 vyskytuje 3x Ala, 6x Val, 1x Ile, používáme tabulku Blosum 62 jaké bude profilové skóre pro výskyt Ile a His ? N(x,A) = 0.3, N(x,V) = 0.6, N(x, I) = 0.1 S(A,I) = -1, S(V,I) = 3, S(I,I) = 4 S(A,H) = -2, S(V,H) = -3, S(I,H) = -3 Prof (x, I) = 0.3 x x x 4 = 2.1 x 10 (v profilu) = 21 ( -1, 3, 4) Prof (x, H) = 0.3 x x x -3 = -2.7 x 10 = -27 (-2, -3, -3)
 = 0.3, N(x,V) = 0.6, N(x, I) = 0.1. S(A,I) = -1, S(V,I) = 3, S(I,I) = 4. S(A,H) = -2, S(V,H) = -3, S(I,H) = -3. Prof (x, I) = 0.3 x x x 4 = 2.1 x 10 (v profilu) = 21 ( -1, 3, 4) Prof (x, H) = 0.3 x x x -3 = -2.7 x 10 = -27 (-2, -3, -3)")
54
PSI-BLAST PSI-BLAST = Position Specific Iterative Blast
Altschul et al., 1997 profilová metoda, používá Position Specific Scoring Matrix (PSSM) v prvním kole klasický BLAST, z vysoko skórujících alignmentů je generována PSSM v dalším kole hledání je už použita nová matrice a následně znovu generována nová PSSM opakováno libovolně dlouho (až ke konvergenci) benchmark metoda
 v prvním kole klasický BLAST, z vysoko skórujících alignmentů je generována PSSM. v dalším kole hledání je už použita nová matrice a následně znovu generována nová PSSM. opakováno libovolně dlouho (až ke konvergenci) benchmark metoda.")
55
HMM HMM = Hidden Markov Model
profilová metoda, používána při rozhodování, zda protein spadá do jisté skupiny proteinů, typicky pro sekvence s nízkou %SI velmi citlivá metoda, která vytváří statistický model pro definovanou skupinu sekvencí na základě “tréninku” na sekvencích patřících do jedné skupiny (globiny) generuje pravděpodobnost nejen pro jednotlivé záměny a inzerce a delece, ale i pro přechody mezi nima dovede do modelu zahrnout i aminokyseliny, které se v tréninkové skupině nevyskytují alignment s největší pravděpodobností je optimální posuzuje jak dobře daná sekvence odpovídá modelu
 generuje pravděpodobnost nejen pro jednotlivé záměny a inzerce a delece, ale i pro přechody mezi nima. dovede do modelu zahrnout i aminokyseliny, které se v tréninkové skupině nevyskytují. alignment s největší pravděpodobností je optimální. posuzuje jak dobře daná sekvence odpovídá modelu.")
56
HMM

57
SHRNUTÍ databáze by měly být pravidelně updatovány
přehled dostupných biologických databází vždy v lednovém čísle NAR řada velmi specializovaných databází hledání v databázích povětšinou heuristickými metodami standard dnes BLAST nutno hodnotit statistickou významnost nálezu citlivější metodou PSI-Blast nebo HMM metody

Podobné prezentace






















































 je většinou myšlenková konstrukce, která obsahuje veškerá data, se kterými pracujeme a není vždy snadné jej.>")

