Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Počítačová část 1. seznámení s on-line databázemi, nástroji a softwarem (databáze, vyhledání sekvencí, základní manipulace se sekvencemi, navržení primerů) 2. fylogenetická analýza
 2. fylogenetická analýza.")
2
začátky chci studovat populační strukturu, paternitu, příbuzenské vztahy uvnitř nějaké skupiny organismů, … podívat se na NCBI a do databází článků (WOS, PubMed) co se o daném ogranismu/ skupině ví jaké geny jsou sekvenovány, jsou na něj známy primery na mikrosatelity? databáze primerů pro mikrosatelity nebo lze hledat ve WOS nebo na stránkách NCBI a zadat klíčová slova: microsatellite AND …(jméno druhu či příbuzných taxonů)
 co se o daném ogranismu/ skupině ví. jaké geny jsou sekvenovány, jsou na něj známy primery na mikrosatelity databáze primerů pro mikrosatelity. nebo lze hledat ve WOS nebo na stránkách NCBI a zadat klíčová slova: microsatellite AND …(jméno druhu či příbuzných taxonů)")
4
NCBI http://www.ncbi.nlm.nih.gov/
celé sekvence mikrosatelitových lokusů Příklad: NCBI Search - Loxia microsatellite Existují i specializované databáze: MGI - příklad DXMit5 (microsatelit na Chr X) Ark - příklad mikrosatelity na Chr Y prasete
 Ark - příklad mikrosatelity na Chr Y prasete.")
5
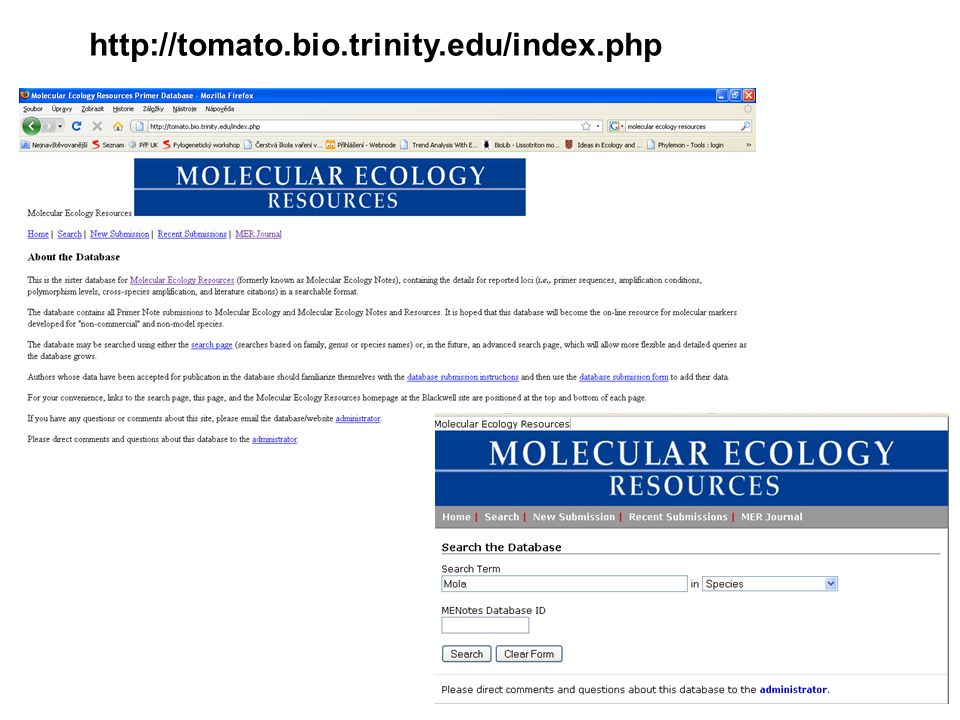
Vyhledávání sekvencí v genové bance (GenBank)
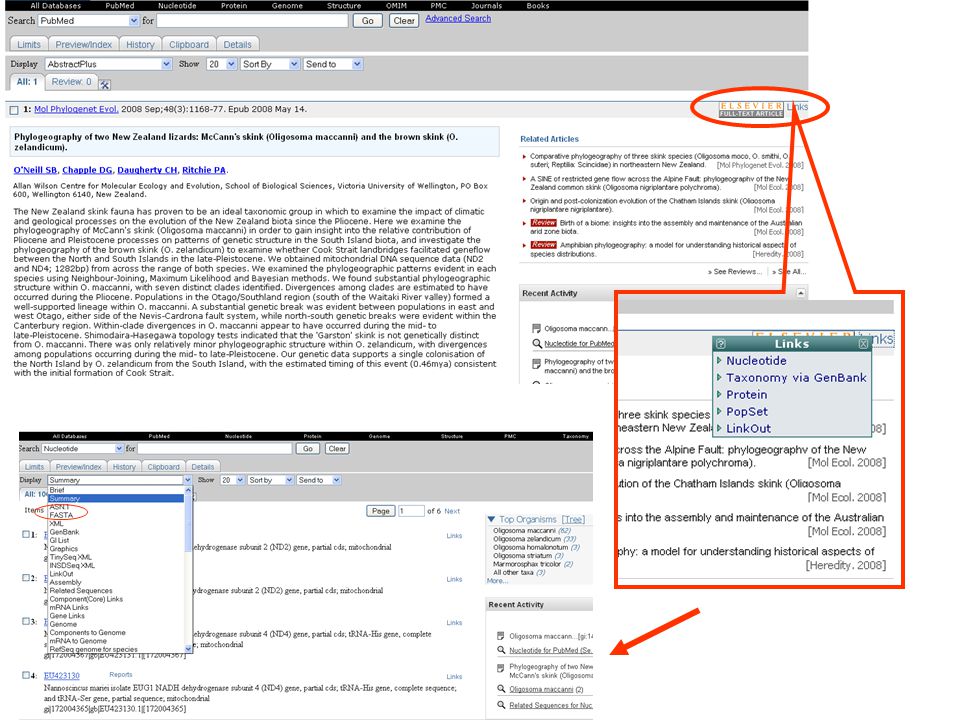
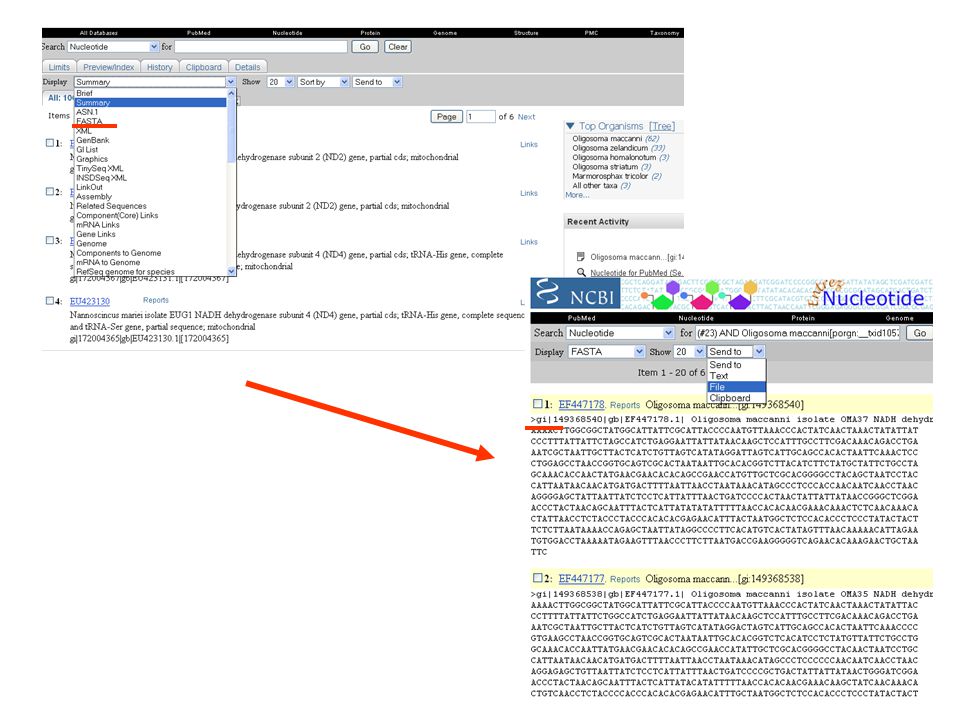
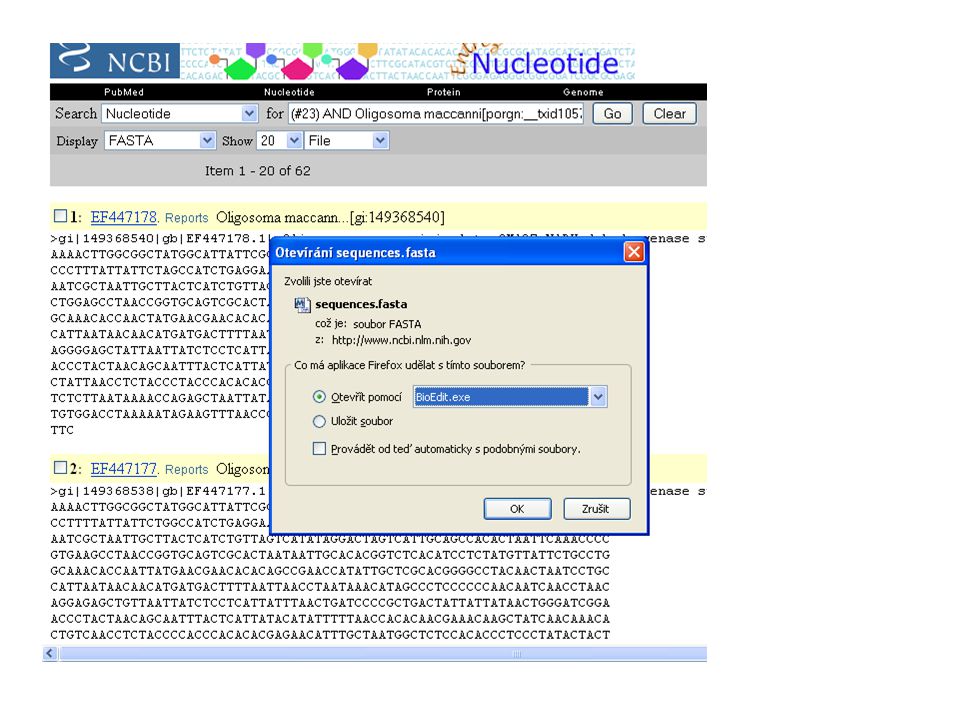
jsou známy nějaké sekvence scinka druhu Oligosoma maccanni ? Chci dělat fylogeografii tohoto druhu – jaké geny použili jiní badatelé? Stažení sekvencí se kterými pracovali přímo v určitém článku snadno a rychle … přes GenBank na NCBI

6
Jak postupovat? V NCBI

10
BioEdit - editor sekvencí - barevné rozlišení - možnost čtení a editace ABI souborů ze sekvenátoru - implementováno mnoho dalších funkcí - alignment - CluslalW - umožňuje slučovat alignmenty, přidávat referenční sekvence, atd. - obsahuje část fylogenetického balíku Phylip - možnost jednoduchých fylogenetických analýz – např. neighbour-joining - BLAST

11
Práce se sekvencemi on-line
SMS- sequence manipulation suite - různá manipulace se sekvencemi, převod formátů, reverse complement, náhodná sekvence, … FaBox- umožňuje práci s datasety, převod formátů, rozpojování a spojování datasetů

12
Kde a čím se sekvence liší?
Alignment - pairwise alignment (dvě sekvence) - multiple alignment (víc sekvencí) alignment start pro fylogenetické analýzy jedná se o ustanovení poziční homologie jednotlivých bází v sekvenci (jednotlivých znaků vstupujících do analýzy) vždy se musí pracovat s homologickými znaky existují různé programy a editory na tvorbu a úpravu alignmentů - ruční BioEdit, Macaw - automatické – pracují s různými algoritmy Clustal X, PileUp, Multalin, Mafft mnoho z těchto programů jsou online
 - multiple alignment (víc sekvencí) alignment start pro fylogenetické analýzy. jedná se o ustanovení poziční homologie jednotlivých bází v sekvenci (jednotlivých znaků vstupujících do analýzy) vždy se musí pracovat s homologickými znaky. existují různé programy a editory na tvorbu a úpravu alignmentů - ruční BioEdit, Macaw. - automatické – pracují s různými algoritmy. Clustal X, PileUp, Multalin, Mafft. mnoho z těchto programů jsou online.")
14
Základy fylogenetických analýz
znaková data (maximální úspornost (parsimony, MP); maximální pravděpodobnost (likelihood, ML), Bayesiánská analýza, BA) dva typy dat distanční data (neighbour-joining, UPGMA) Přístup metod k výpočtu fylogenetického stromu je dvojí: používají k výpočtu algoritmus (sled specifických kroků) nebo nějaké kritérium optimálnosti MP - princip metody je vybrat strom s minimální celkovou délkou (nejmenší počtem evolučních kroků – např. nejméně substitucí nukleotidů) , metodou se často nalezne několik stejně parsimonních stromů - pro získání jednoho stromu – nutno udělat konsenzuální strom ML – metoda posuzuje hypotézy o evoluční historii zkoumaných taxonů z hlediska pravděpodobnosti, že jsou v souladu se získanými daty. Vyšší pravděpodobnost stromu je preferována nad nižší, nutno zadat model evoluce sekvencí.
; maximální pravděpodobnost (likelihood, ML), Bayesiánská analýza, BA) dva typy dat. distanční data (neighbour-joining, UPGMA) Přístup metod k výpočtu fylogenetického stromu je dvojí: používají k výpočtu algoritmus (sled specifických kroků) nebo nějaké kritérium optimálnosti. MP - princip metody je vybrat strom s minimální celkovou délkou (nejmenší počtem evolučních kroků – např. nejméně substitucí nukleotidů) , metodou se často nalezne několik stejně parsimonních stromů - pro získání jednoho stromu – nutno udělat konsenzuální strom. ML – metoda posuzuje hypotézy o evoluční historii zkoumaných taxonů z hlediska pravděpodobnosti, že jsou v souladu se získanými daty. Vyšší pravděpodobnost stromu je preferována nad nižší, nutno zadat model evoluce sekvencí.")
15
Základy fylogenetických analýz
ML – vychází z modelu evolučního procesu, který vede ke změně jedné sekvence v druhou (substituční model) často se model navrhuje pomocí dalších programů (Modeltest) na základě vstupních dat - obecně modely uvažují: frekvenci jednotlivých bází pravděpodobnost změny jednoho nukleotidu v druhý (transice x transverze) heterogenita substitučních rychlostí v různých částech sekvence příklady modelů: Jukes-Cantor, Kimurův dvouparametrový model, general time-reversible model Výsledkem ML je jeden strom Další znakovou metodou, založenou na modelu evoluce sekvencí a používající při výpočtu posteriorní pravděpodobnosti (probability) - je Bayesovská analýza (BA) Distanční metody: př.Neighbour-joining (NJ) - používají genetické distance, používá korekce distancí, aby umožnila odhadnout počet nezjištěných změn (podobně jako substituční modely ML)
 často se model navrhuje pomocí dalších programů (Modeltest) na základě vstupních dat - obecně modely uvažují: frekvenci jednotlivých bází. pravděpodobnost změny jednoho nukleotidu v druhý (transice x transverze) heterogenita substitučních rychlostí v různých částech sekvence. příklady modelů: Jukes-Cantor, Kimurův dvouparametrový model, general time-reversible model. Výsledkem ML je jeden strom. Další znakovou metodou, založenou na modelu evoluce sekvencí a používající při výpočtu posteriorní pravděpodobnosti (probability) - je Bayesovská analýza (BA) Distanční metody: př.Neighbour-joining (NJ) - používají genetické distance, používá korekce distancí, aby umožnila odhadnout počet nezjištěných změn (podobně jako substituční modely ML)")
16
podpora zjištěných topologií (stanovení spolehlivosti)
nejčastěji využití neparametrických technik opakovaného výběru bootstrapping, jackknifing náhodné vybírání znaků z původního datasetu (nukleotidů) ve fylogenetice vlastně vytvoříme mnoho datasetů (založených na stejných datech jako originální dataset, ale v jiném zastoupení), z každého datasetu vypočteme strom, ze všech takto vzniklých stromů vytvoříme konsenzuální strom a procentuální zastoupení jednotlivých větvení (topologie taxonu) ukazuje na míru spolehlivosti větve s bootstrapem < 50% - nelze topologii věřit – může se jednat o náhodu, nad 75% u MP, ML – uspokojivě spolehlivé, % velmi dobré, pozor na BA – zde se jedná o posteriorní probability- pod 95 (resp. 0.95) topologie nejistá
 ve fylogenetice vlastně vytvoříme mnoho datasetů (založených na stejných datech jako originální dataset, ale v jiném zastoupení), z každého datasetu vypočteme strom, ze všech takto vzniklých stromů vytvoříme konsenzuální strom a procentuální zastoupení jednotlivých větvení (topologie taxonu) ukazuje na míru spolehlivosti. větve s bootstrapem < 50% - nelze topologii věřit – může se jednat o náhodu, nad 75% u MP, ML – uspokojivě spolehlivé, % velmi dobré, pozor na BA – zde se jedná o posteriorní probability- pod 95 (resp. 0.95) topologie nejistá.")
17
Take -Home Message! Software: MP: PAUP*, TNT, Phylip, …
celkem je několik metod rekonstrukce fylogenetických stromů fylogenetický strom jako výstup jakékoliv metody je jen hypotéza - musíme zjistit spolehlivost topologií výsledek vždy závisí na kvalitě vstupních dat a na dobrém alignmentu (určení homologických znaků, které pak porovnáváme) Software: MP: PAUP*, TNT, Phylip, … ML: PAUP*, PHYML, GARLI, RAxML, Phylip BA: MrBayes NJ: PAUP*, Phylip, MEGA, … + různé internetové aplikace
 Software: MP: PAUP*, TNT, Phylip, … ML: PAUP*, PHYML, GARLI, RAxML, Phylip. BA: MrBayes. NJ: PAUP*, Phylip, MEGA, … + různé internetové aplikace.")
18
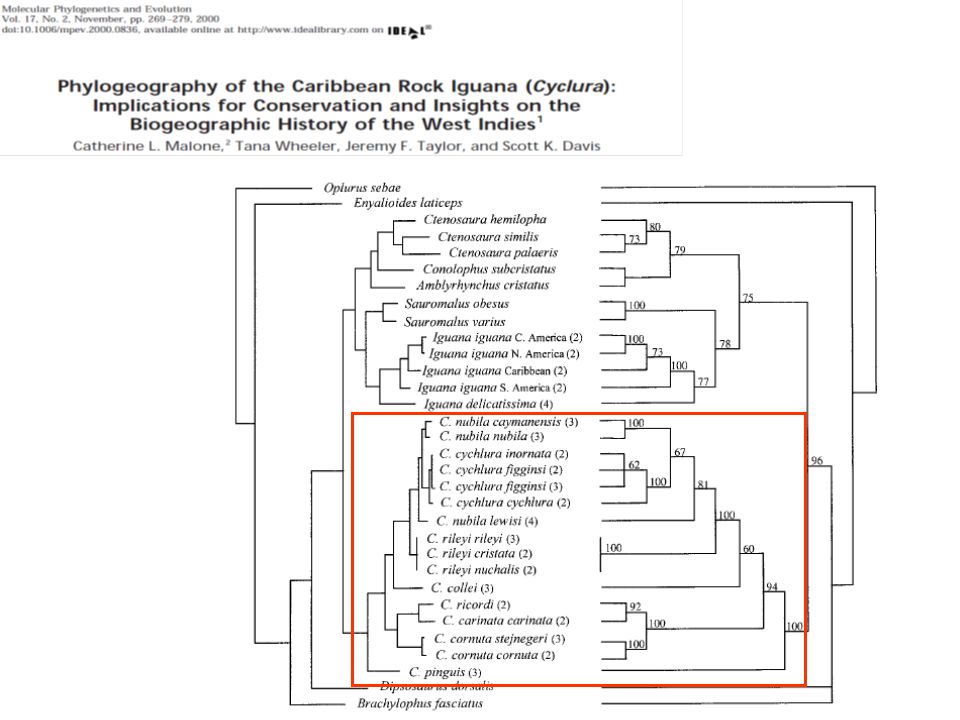
úloha: příbuzenské vztahy leguánů – zejm. rod Cyclura stáhneme sekvence z GenBank – pro tento případ si stáhneme sekvence použité Malone et al Phylogeography of the Caribbean rock iguana (Cyclura): implications for conservation and insights on the biogeographic history of the West Indies. stáhnout sekvence použité autory do souboru –FASTA formát – otevřít v BioEdit - pozměnit jména aby byla zpracovatelná programy a srozumitelná (8 znaků) udělat alignment - Accesory Application - ClustalW vytvořit fylogenetický strom metodou NJ a Maximální parsimonie v BioEditu pomocí Accesory Application – DNApars, … - outfile a outtree
: implications for conservation and insights on the biogeographic history of the West Indies. stáhnout sekvence použité autory. do souboru –FASTA formát – otevřít v BioEdit - pozměnit jména aby byla zpracovatelná programy a srozumitelná (8 znaků) udělat alignment - Accesory Application - ClustalW. vytvořit fylogenetický strom metodou NJ a Maximální parsimonie v BioEditu pomocí Accesory Application – DNApars, … - outfile a outtree.")
19
Strom v závorkové konvenci – lze vložit např
Strom v závorkové konvenci – lze vložit např. do TreeView a zobrazit v grafické podobě (IguigNA1_: ,(Iguig: ,(((Cpinguis: ,((((Cstej1: , Cstej2: ): ,Ccor: ): ,((Cric1: , Cric2: ): ,(Cyccar2: ,Cyccar1: ): ): ): , (Ccollei: ,(Crileyi: ,((Clew1: ,Clew2: ): , (((Ccay1: ,Ccay2: ): ,(Cnubnub1: ,Cnubnub2: ): ): ,((Cinor: ,Cfig1: ,Cfig2: ): ,(Ccychc2: , Ccychc: ): ): ): ): ): ): ): ): ,Igudeli: ): ,IguigSA1_: ): ): ,IguigCA1_: ); stromy lze prohlížet v různých programech TreeView, FigTree, Dendroscope
: ,Ccor: ): ,((Cric1: , Cric2: ): ,(Cyccar2: ,Cyccar1: ): ): ): , (Ccollei: ,(Crileyi: ,((Clew1: ,Clew2: ): , (((Ccay1: ,Ccay2: ): ,(Cnubnub1: ,Cnubnub2: ): ): ,((Cinor: ,Cfig1: ,Cfig2: ): ,(Ccychc2: , Ccychc: ): ): ): ): ): ): ): ): ,Igudeli: ): ,IguigSA1_: ): ): ,IguigCA1_: ); stromy lze prohlížet v různých programech TreeView, FigTree, Dendroscope.")
21
Kde hledat na internetu?
stránky J. Felsensteina: -téměř úplný přehled programů pro fylogenetické analýzy, zobrazování stromů, testy, a vše další potřebné portál Phylemon: -některé analýzy on-line

22
Vyhledání restrikčních míst
Webcutter

23
Vyhledání restrikčních míst Myš, mtDNA, restrikční místo na pozici 3565 pro BamHI Restrikční místo je jen u jednoho z poddruhů.

24
Potřebujeme vyhledat primery
Stáhnutí sekvence (znám Acc. No. z článku) Ověřit, zda sekvence obsahuje restrikční místo Vyhledání primerů pro amplifikaci úseku, který obsahuje restrikční místo Restrikční místo GGATTC Primer Primer CCTAAG bp
 Ověřit, zda sekvence obsahuje restrikční místo. Vyhledání primerů pro amplifikaci úseku, který obsahuje restrikční místo. Restrikční místo. GGATTC. Primer. Primer. CCTAAG bp.")
25
Vše přes Internet Sekvence NCBI Primery PRIMER3
Restrikční místo Webcutter

26
Restrikční místo v mtND1
ACCESSION NC_005089 Restrikční místo 3565 pro BamHI

27
Kde se dozvědět více? Kurz Computational Genomics (Marc VanRanst, Bioinformatics bookmarks Introduction to Bioinformatics (F. Cvrčková, Molekulární ekologie (Pavel Munclinger, letní semestr, populační genetika, analýza paternity) Evoluční genetika (Pavel Munclinger a Radka Storchová, zimní semestr) Molekulární taxonomie + cvičení (J. Flégr, cvičení V. Hampl Fylogenetický workshop (
 Evoluční genetika. (Pavel Munclinger a Radka Storchová, zimní semestr) Molekulární taxonomie + cvičení. (J. Flégr, cvičení V. Hampl Fylogenetický workshop. (")
28
Kde najdu adresy stránek z tohoto praktika?
( Další čtení: Phylogenetic Trees Made Easy: A How-To Manual, Third Edition Barry G. Hall, Emeritus, University of Rochester Genetické metody v zoologii J. Zima, M. Macholán, P. Munclinger, J. Piálek Karolinum 2004 Úvod do praktické bioinformatiky F. Cvrčková Academia

29
Na GenBank najít sekvenci cytochromu b slona (Elephas), zkopírovat
úloha: Na GenBank najít sekvenci cytochromu b slona (Elephas), zkopírovat jí ve FASTA formátu > jméno sekvence ATGTGCTTGGAATCCAT do poznámkového bloku a pak: udělat její komplementární sekvenci (např. SMS) převést do proteinů (např. SMS, BioEdit) >gi| |gb|AY | Elephas maximus clone ABC 6 cytochrome b (cytb) gene, complete cds; mitochondrial ATGACCCACACCCGAAAATCTCACCCCCTGTTTAAAATCATCAACAAATCCTTCATTGATCTACCCACCC CATCTAACATCTCAACATGATGAAATTTCGGCTCACTACTAGGAGCGTGCCTAATTACCCAAATCCTAAC AGGATTATTCCTAGCCATACATTACACACCTGACACAATAACTGCATTTTCATCTATATCCCATATCTGC CGAGACGTCAACTACGGCTGAATTATTCGACAACTGCACTCAAACGGAGCATCTATCTTTTTCCTCTGCC TATACACACACATTGGACGAAACATCTACTATGGATCCTATCTATACTCAGAAACCTGAAACACAGGTAT TATACTACTACTAATCACCATAGCCACCGCCTTCATAGGATATGTCCTTCCATGAGGACAAATATCATTC TGAGGGGCAACCGTAATTACTAACCTCTTCTCAGCAATTCCCTACATCGGCACAAACCTAGTAGAATGAA TTTGAGGAGGCTTTTCGGTAGATAAAGCAACCTTAAACCGATTCTTCGCCTTCCATTTCATCCTTCCATT TACTATAGTTGCACTAGCAGGAGTGCACCTAACCTTTCTTCACGAAACAGGCTCAAACAACCCACTAGGT CTCACTTCAGACTCAGACAAAATTCCCTTCCACCCGTACTATACTATCAAAGACTTCCTAGGACTACTTA TCCTAATTTTACTCCTTCTACTCTTAGCCCTACTATCTCCAGACATACTAGGAGACCCTGACAACTACAT ACCAGCTGATCCACTAAATACCCCCCTACATATCAAACCAGAGTGATACTTCCTTTTTGCTTACGCCATC CTACGATCTGTACCAAATAAACTAGGAGGCGTCCTAGCCCTACTCCTATCAATCCTAATTTTAGGATTAA TACCACTTCTCCACACATCCAAACATCGAAGTATAATACTCCGACCTCTCAGCCAAGTCCTATTCTGAGC TCTAACAATAGACCTACTAATACTTACATGAATTGGCAGCCAACCAGTAGAATATCCTTACATCGCTATC GGTCAAATAGCCTCAATTCTATACTTCTCTATTATTCTAGCCTTCCTACCAATTGCAGGAATAATCGAAAA CTACCTCATTAAATAA
, zkopírovat. jí ve FASTA formátu > jméno sekvence. ATGTGCTTGGAATCCAT. do poznámkového bloku a pak: udělat její komplementární sekvenci (např. SMS) převést do proteinů (např. SMS, BioEdit) >gi| |gb|AY | Elephas maximus clone ABC 6 cytochrome b (cytb) gene, complete cds; mitochondrial. ATGACCCACACCCGAAAATCTCACCCCCTGTTTAAAATCATCAACAAATCCTTCATTGATCTACCCACCC CATCTAACATCTCAACATGATGAAATTTCGGCTCACTACTAGGAGCGTGCCTAATTACCCAAATCCTAAC AGGATTATTCCTAGCCATACATTACACACCTGACACAATAACTGCATTTTCATCTATATCCCATATCTGC CGAGACGTCAACTACGGCTGAATTATTCGACAACTGCACTCAAACGGAGCATCTATCTTTTTCCTCTGCC TATACACACACATTGGACGAAACATCTACTATGGATCCTATCTATACTCAGAAACCTGAAACACAGGTAT TATACTACTACTAATCACCATAGCCACCGCCTTCATAGGATATGTCCTTCCATGAGGACAAATATCATTC TGAGGGGCAACCGTAATTACTAACCTCTTCTCAGCAATTCCCTACATCGGCACAAACCTAGTAGAATGAA TTTGAGGAGGCTTTTCGGTAGATAAAGCAACCTTAAACCGATTCTTCGCCTTCCATTTCATCCTTCCATT TACTATAGTTGCACTAGCAGGAGTGCACCTAACCTTTCTTCACGAAACAGGCTCAAACAACCCACTAGGT CTCACTTCAGACTCAGACAAAATTCCCTTCCACCCGTACTATACTATCAAAGACTTCCTAGGACTACTTA TCCTAATTTTACTCCTTCTACTCTTAGCCCTACTATCTCCAGACATACTAGGAGACCCTGACAACTACAT ACCAGCTGATCCACTAAATACCCCCCTACATATCAAACCAGAGTGATACTTCCTTTTTGCTTACGCCATC CTACGATCTGTACCAAATAAACTAGGAGGCGTCCTAGCCCTACTCCTATCAATCCTAATTTTAGGATTAA TACCACTTCTCCACACATCCAAACATCGAAGTATAATACTCCGACCTCTCAGCCAAGTCCTATTCTGAGC TCTAACAATAGACCTACTAATACTTACATGAATTGGCAGCCAACCAGTAGAATATCCTTACATCGCTATC GGTCAAATAGCCTCAATTCTATACTTCTCTATTATTCTAGCCTTCCTACCAATTGCAGGAATAATCGAAAA. CTACCTCATTAAATAA.")
30
úloha: Ze sekvencí leguánů z předchozího příkladu (ve formátu FASTA)
udělejte za použití metody maximum likelihood (program Phyml) fylogenetický strom návod: Na stránkách FABOX si převedeme data ve formátu FASTA do vstupního formátu pro program PAML (stejný jako vstupní formát pro program Phyml, který budeme používat) Create formatted sequence file for PAML analysis (fasta2paml) uložit soubor otevřít nový soubor *.phy – prohlédnout si strukturu formátu počet sekvencí v datasetu délka sekvence název vzorku vlastní sekvence ve formátu interleaved
 fylogenetický strom. návod: Na stránkách FABOX si převedeme data ve formátu FASTA do vstupního formátu pro program PAML (stejný jako vstupní formát pro program Phyml, který budeme používat) Create formatted sequence file for PAML analysis (fasta2paml) uložit soubor otevřít nový soubor *.phy – prohlédnout si strukturu formátu. počet sekvencí. v datasetu. délka sekvence. název vzorku. vlastní sekvence. ve formátu interleaved.")
31
portál Phylemon volba Phyml – nahrajeme vstupní soubor *.phy a zadáme parametry analýzy (viz obr.) zadáme nejkomplexnější model evoluce sekvence (GTR) a topologii větví ověříme bootstrapem se 100 pseudoreplikacemi, ostatní parametry necháme určit programem na portálu je možné i vizualizovat konečný strom nebo si výstupní strom pouze uložit
 a topologii větví ověříme bootstrapem se 100 pseudoreplikacemi, ostatní parametry necháme určit programem. na portálu je možné i vizualizovat konečný strom. nebo si výstupní strom pouze uložit.")
Podobné prezentace

















 2. Fylogenetická analýza.>")

