Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
10. Čistící a sanitační procesy

2
Čistící a sanitační procesy Klasifikace farmaceutických látek z hlediska nebezpečných účinků na personál ve výrobě JKTP 2013

3
Základní pojmy Validace je dokumentované ověření, že proces nebo jeho část standardně dosahuje specifikované parametry Kvalifikace je dokumentované ověření, že zařízení nebo jeho část standardně dosahuje specifikované parametry. Kontaminace je biologické, chemické nebo fyzikální znečištění výchozí látky, obalového materiálu, meziproduktu, nerozplněného produktu nebo konečného produktu. JKTP 2013

4
Kontaminace vyjadřuje množství nečistot v prostředí, zařízení nebo materiálu. Křížová kontaminace je znečištění výchozí látky nebo produktu jinou výchozí látkou, obalovým materiálem nebo produktem. Ke křížové kontaminaci může dojít: Nedokonale čistým výrobním zařízením Nedokonalou čistotou vzduchu ve výrobním prostoru Nevhodným chováním obsluhujícího, příp. jiného personálu JKTP 2013

5
ČISTOTA Čistota je definována jako úroveň kontaminace, která je nižší než určitá limitní koncentrace nečistot. JKTP 2013

6
Validace čistících postupů Cílem je prokázat, že při důsledném používání validovaných čistících postupů se během výroby léčiv neprojeví problémy spojené s jejich kontaminací. Provádí se za účelem potvrzení účinnosti čistících postupů. JKTP 2013

7
PROČ SE FARMACEUTICKÉ FIRMY VĚNUJÍ VALIDACÍM ČISTÍCÍHO POSTUPU ? Co je důvodem, že každý rok vynakládají obrovské finanční prostředky na to, aby mohly svým zákazníkům, příp. auditorům doložit, že jejich čistící postupy fungují naprosto bezchybně a že jejich výrobky nejsou křížově kontaminovány a co se týká čistoty dosahují tudíž naprosto perfektní kvality? Nebylo by dostačující důsledně provádět vizuální kontrolu čistoty s vědomím, že člověk je schopen rozpoznat nečistotu na úrovni 400 µg/plochu? JKTP 2013

8
Tyto důvody můžeme vysvětlit na následujícím příkladu Farmaceutická firma VW má ve svém sortimentu pevných lékových forem dva přípravky, které vyrábí na stejných zařízeních po sobě: Přípravek ABC (skupina laxativ), velmi nízká denní terapeutická dávka Přípravek XYZ (skupina psychofarmak), vysoká denní terapeutická dávka JKTP 2013
, velmi nízká denní terapeutická dávka Přípravek XYZ (skupina psychofarmak), vysoká denní terapeutická dávka JKTP 2013")
9
V důsledku toho, že mycí procesy neprobíhají ve firmě VW zcela dokonale a přestože výrobní zařízení jsou po ukončení mycího procesu vizuálně naprosto čisté, může dojít k situaci, že v maximální denní dávce přípravku XYZ je taková kontaminace přípravkem ABC, která se rovná minimální denní dávce tohoto přípravku. Příklad: Pan Vomáčka JKTP 2013

11
HISTORIE R. 1988, FDA (Food and drug administration) – křížová kontaminace způsobená nedostatečným čištěním a ověřením Výrobce ú.l. Cholestyramine Resin USP stahoval z oběhu produkt v důsledku vysokého obsahu zemědělských pesticidů Původ kontaminace: použití kontejnerů v chemické výrobě, které byly použity i pro výrobu zemědělských pesticidů. Kontejnery nebyly dostatečně vyčištěny Tato událost zvýšila povědomí FDA v souvislosti s možností vzniku křížové kontaminace a varovala chemické a farmaceutické firmy před budoucím požadavkem na ověřování (validací) čistícího procesu JKTP 2013
 – křížová kontaminace způsobená nedostatečným čištěním a ověřením Výrobce ú.l. Cholestyramine Resin USP stahoval z oběhu produkt v důsledku vysokého obsahu zemědělských pesticidů Původ kontaminace: použití kontejnerů v chemické výrobě, které byly použity i pro výrobu zemědělských pesticidů. Kontejnery nebyly dostatečně vyčištěny Tato událost zvýšila povědomí FDA v souvislosti s možností vzniku křížové kontaminace a varovala chemické a farmaceutické firmy před budoucím požadavkem na ověřování (validací) čistícího procesu JKTP")
12
ČISTÍCÍ POSTUPY Manuální /tabletovací stroje, kapslovací stroje, blistrovací stroje/ Poloautomatické /některé homogenizační kotle ve výrobě PTLF/ Automatické (CIP) /fluidní sušárny a granulátory JKTP 2013
 /fluidní sušárny a granulátory JKTP 2013")
13
Předpokladem pro provádění validací čistících postupů jsou Zpracovaná SOP detailně popisující průběh čistícího procesu - mají být jednoznačná a srozumitelná - mají definovat čistící prostředek, jeho množství a koncentraci, jednotlivé čistící kroky, jejich dobu trvání, kvalitu a teplotu oplachové vody, specifikovat čistící nářadí a případně stanovit čas, do kdy je třeba čištění provést po ukončení výroby JKTP 2013

14
Operátoři - vyškolení z příslušného čistícího SOP - dobře trénovaní, rozdíly mezi jejich odvedenou prací musí být minimální Čistící postupy musí být - pod kontrolou - dokumentované, kdo a kdy je provedl a kontroloval JKTP 2013

15
Princip validace čistícího postupu Zařízení nebo předměty se myjí podle daného postupu, který je uveden v SOP. Po úspěšném dokončení mycího procesu je zvolenou metodou odebraný vzorek. Odběr vzorku se provádí po třech mycích cyklech. Vzorky se analyticky vyhodnotí a porovnají se se stanoveným kriteriem přijatelnosti po korekci na zjištěný Recovery factor. Jako sledovaná látka je zvolená účinná látka, detergent nebo látka pomocná, která je z hlediska čistitelnosti významná (těžko čistitelná, nebezpečí křížové kontaminace z důvodů chemických resp. fyzikálních vlastností – pH, barevnost). JKTP 2013
. JKTP")
16
Princip metody Procesní kvalifikace čistícího postupu je prováděná následujícími metodami: - vizuální kontrola čistoty - analýza stěrů reziduí sledované látky - analýza poslední oplachové vody, resp. posledního oplachového media, měření vodivosti - mikrobiologické hodnocení stěrů z povrchu zařízení nebo poslední oplachové vody JKTP 2013

17
Zásady pro plánování procesní kvalifikace čistícího postupu Konkrétní zkoušky pro kalendářní rok jsou uvedené ve VMP. Procesní kvalifikace čistícího postupu je plánována pro nově zaváděné účinné látky, pro nové zařízení, pro nové systémy CIP, pro nově používané detergenty. Procesní kvalifikaci čistícího postupu není třeba plánovat u vitaminových přípravků, pokud k tomu nejsou závažné důvody (obtížná čistitelnost,..) Pro výrobní linku, příp. výrobní zařízení je možné na základě metody určení „nejhoršího případu“ zvolit přípravek, který zastupuje všechny na této výrobní lince zpracovávané přípravky (princip slučování přípravků do skupin). Je též možné použít metody založené na principu slučování podobných čistících postupů, resp. podobnosti výrobních zařízení. JKTP 2013
 Pro výrobní linku, příp. výrobní zařízení je možné na základě metody určení „nejhoršího případu zvolit přípravek, který zastupuje všechny na této výrobní lince zpracovávané přípravky (princip slučování přípravků do skupin). Je též možné použít metody založené na principu slučování podobných čistících postupů, resp. podobnosti výrobních zařízení. JKTP")
18
Zásady pro plánování procesní kvalifikace čistícího postupu Při hodnocení nejhoršího případu se postupuje podle následujícich kritérií: a) Obtížnost čištění (praktické zkušenosti) b) Rozpustnost v čistícím médiu c) Největší toxicita d) Nejmenší terapeutická dávka e) Nejmenší limit ( z terapeutické dávky, toxik. dat,.......) Jednotlivé parametry se bodově ohodnotí a provede se vyhodnocení a určení nejhoršího případu. Rovněž je třeba přihlédnout k frekvenci výroby. Způsob výběru nejhoršího případu: Jednotlivá kriteria se bodově ohodnotí. Za předpokladu, že všechny kriteria mají stejnou váhu (důležitost), výsledek s nejvyšším bodovým hodnocením udává nejhorší případ v rámci dané skupiny. JKTP 2013
 Jednotlivé parametry se bodově ohodnotí a provede se vyhodnocení a určení nejhoršího případu. Rovněž je třeba přihlédnout k frekvenci výroby. Způsob výběru nejhoršího případu: Jednotlivá kriteria se bodově ohodnotí. Za předpokladu, že všechny kriteria mají stejnou váhu (důležitost), výsledek s nejvyšším bodovým hodnocením udává nejhorší případ v rámci dané skupiny. JKTP")
19
Zásady pro realizaci procesní kvalifikace čistícího postupu Výrobní zařízení – umyté podle postupu v SOP. Následná vizuální kontrola. Provedení odběru vzorku z vytipovaných kritických míst. Určení odběrových míst – jsou definované standardně pro jednotlivé typy zařízení v SOP a následně ve VP pro každý přípravek. Stěr je odebrán např. polyesterovou stěrovou tyčinkou nebo vatovým smotkem (upraveno extrakcí v methanolu). Stěrová tyčinka je navlhčena kapalinou, která je vhodným rozpouštědlem sledované látky. Je žádoucí, aby sledovaná látka byla v rozpouštědle velmi dobře rozpustná. JKTP 2013
. Stěrová tyčinka je navlhčena kapalinou, která je vhodným rozpouštědlem sledované látky. Je žádoucí, aby sledovaná látka byla v rozpouštědle velmi dobře rozpustná. JKTP")
20
Zásady pro realizaci procesní kvalifikace čistícího postupu Plocha stěru - optimálně má rozměr 10x10 cm. Při každé sérii stanovení se připraví současně k odebraným vzorkům slepý vzorek. Hlavičky tyčinek se vloží do skleněných vzorkovnic. Rozpouštědlo je možné nechat samovolně odpařit (podle závěrů validace analytické metody). Vzorkovnice a jejich uzávěry nesmí uvolňovat rezidua, které by mohly ovlivnit výsledek analýzy. Takto připravený vzorek se předává do analytické laboratoře. (společně s protokolem o předání vzorků). Laboratoř stanoví obsah sledované látky v µg na jeden stěr validovanou analytickou metodou. JKTP 2013
. Vzorkovnice a jejich uzávěry nesmí uvolňovat rezidua, které by mohly ovlivnit výsledek analýzy. Takto připravený vzorek se předává do analytické laboratoře. (společně s protokolem o předání vzorků). Laboratoř stanoví obsah sledované látky v µg na jeden stěr validovanou analytickou metodou. JKTP")
21
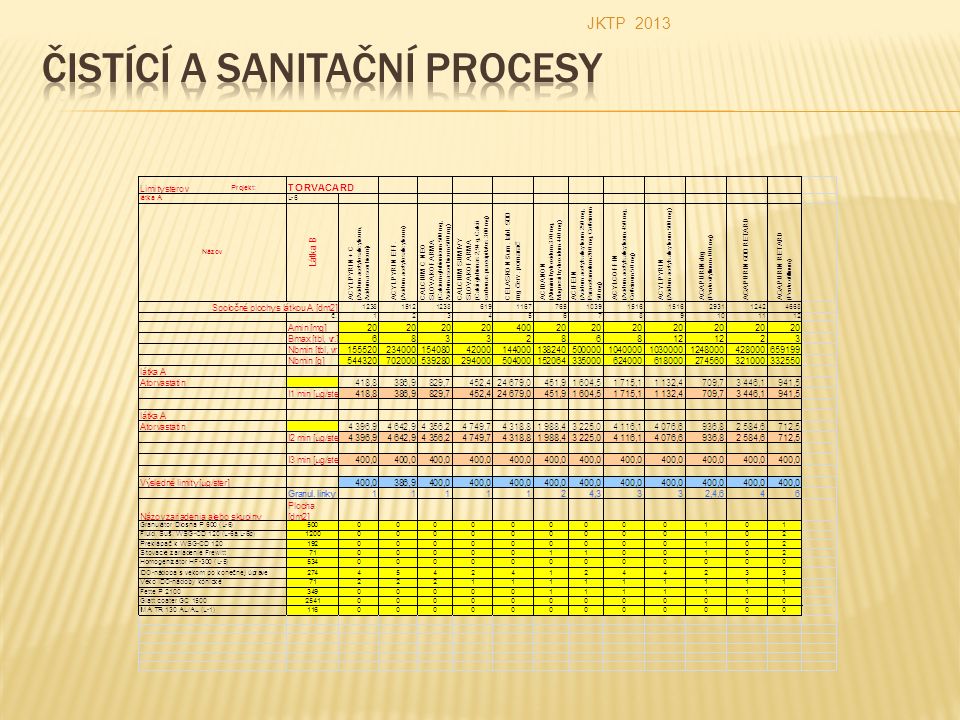
Kriteria přijatelnosti procesní kvalifikace čistícího postupu Pro výpočet hodnoty maximálně přípustného znečištění jsou definovány limity I 1, I 2, I 3 - pro stěry I - pro oplachy Porovnáním hodnot (platí pro stěry) - je určen limit s nejnižší hodnotou jako tzv. výsledný limit (kriterium přijatelnosti). JKTP 2013
. JKTP")
22
Kriteria přijatelnosti procesní kvalifikace čistícího postupu Limit I 1 Vychází z požadavku, že v maximální terapeutické dávce přípravku B se nesmí objevit více než 1/1000 minimální terapeutické denní dávky účinné látky z přípravku A. Lze jej použít pouze tehdy, je – li sledovanou (kontaminující) látkou účinná látka s definovanou terapeutickou denní dávkou. Pokud předpokládáme: FN = 10, FB = 10 a FV = 10, (FN,FB,FV = bezpečnostní faktory) po zavedení SS = 1 dm 2 (plocha stěru) A min budeme dosazovat v /mg/ (minimální terapeutická dávka látky A) B max (maximální dávka přípravku B) a NB min (minimální velikost zpracovávané šarže přípravku B) budeme dosazovat v /g resp. počtu tablet /, a S AB (celková pracovní plocha výrobní linky) budeme dosazovat v /dm 2 /, dostaneme: JKTP 2013
 látkou účinná látka s definovanou terapeutickou denní dávkou. Pokud předpokládáme: FN = 10, FB = 10 a FV = 10, (FN,FB,FV = bezpečnostní faktory) po zavedení SS = 1 dm 2 (plocha stěru) A min budeme dosazovat v /mg/ (minimální terapeutická dávka látky A) B max (maximální dávka přípravku B) a NB min (minimální velikost zpracovávané šarže přípravku B) budeme dosazovat v /g resp. počtu tablet /, a S AB (celková pracovní plocha výrobní linky) budeme dosazovat v /dm 2 /, dostaneme: JKTP")
23
Kriteria přijatelnosti procesní kvalifikace čistícího postupu Limit I 2 Limit I 2 vychází z požadavku, že v přípravku B se může objevit 10 ppm účinné látky přípravku A. Vzorec pro přípustné množství látky A nalezené ve stěru je potom Pokud předpokládáme: NL = 10 ppm (koncentrace sledované látky) pokud použijeme stejně jako pro I 1 SS = 1dm 2, NB min v /g/ a S AB v /dm 2 /, dostaneme JKTP 2013
 pokud použijeme stejně jako pro I 1 SS = 1dm 2, NB min v /g/ a S AB v /dm 2 /, dostaneme JKTP")
24
Kriteria přijatelnosti procesní kvalifikace čistícího postupu Limit I 3 Hodnota tohoto limitu vychází z požadavku, aby na umytém povrchu zařízení nebylo vizuálně patrné znečištění. Limity I 1 a I 2 bývají u řady přípravků splněny při takových úrovních znečištění povrchu, při kterých jsou rezidua po mytí ještě vizuálně patrná. Z hlediska požadavků SVP je nepřijatelné, aby zařízení s vizuálními nečistotami na vnitřním povrchu bylo označeno jako čisté. V takovém případě je nutno v mytí pokračovat dokud není dosaženo vizuální čistoty povrchu. Studie prokázaly, že účinné látky většiny přípravků jsou vizuálně ještě pozorovatelné na umytém povrchu při koncentracích větších než asi 400 /µg/dm 2 /. Tato hodnota (jestliže je plocha 1 stěru = 1 dm 2 ) byla použita pro číselné vyjádření vizuální čistoty povrchu I 3 = 400 / µg/stěr / U topických lékových forem, u nichž není uvedena hodnota minimální a maximální denní terapeutické dávky, se porovnávají pouze hodnoty I 2 a I 3. Porovnáním všech tří, resp. dvou limitů je určen limit s nejnižší hodnotou jakožto tzv. výsledný limit. JKTP 2013
 byla použita pro číselné vyjádření vizuální čistoty povrchu I 3 = 400 / µg/stěr / U topických lékových forem, u nichž není uvedena hodnota minimální a maximální denní terapeutické dávky, se porovnávají pouze hodnoty I 2 a I 3. Porovnáním všech tří, resp. dvou limitů je určen limit s nejnižší hodnotou jakožto tzv. výsledný limit. JKTP")
25
Příklad Ve firmě Zentiva jsou vyráběny přípravky Neurol tbl. a Ibalgin tbl.flm. na stejných výrobních zařízeních. Úkol: ověřit, zda-li čistící proces zajišťuje dokonalé odstranění přípravku Neurol tbl., resp. vyloučit křížovou kontaminaci přípravku Ibalgin tbl.flm. přípravkem Neurol tbl. Vyhodnocovat rezidua účinné látky Charakteristika vyráběných přípravků Neurol tbl., anxiolytikum úč. látka: Alprazolam, psychotropní látka, seznam IV dle ČL 2009 obsah v tbl. = 0,25 mg použití: úzkostné neurozy, panické stavy, fobie, neurotické deprese …….. způsob výroby: metoda suché granulace, přímé lisování Ibalgin tbl.flm., antirevmatikum, analgetikum úč. látka: Ibuprofen obsah v tbl.flm. = 200 mg použití: zánětlivé degenerativní formy postižení periferních kloubů, horečnaté stavy při zánětech horních cest dýchacích, bolesti způsob výroby: metoda mokré granulace, hnětením JKTP 2013

26
Stanovit kriterium přijatelnosti této zkoušky A min. - Minimální terapeutická dávka látky A NB min. - Minimální velikost zpracovávané šarže přípravku B B max. - maximální terapeutická denní dávka přípravku B S AB - celková plocha těch povrchů technologických zařízení, které přicházejí do kontaktu s látkou A i látkou B JKTP 2013

27
Stanovit kriterium přijatelnosti této zkoušky A min. = 0,25 mg NB min. = 300 000 g B max. = 1,8 g, resp. 2,808 g (přepočteno na hmotnost tablet, 9 tablet o obsahu 200 mg v tabletě) S AB = 3100 dm 2 I 1 = 8,6 µg/stěr ( plocha stěru = 1 dm 2 ) Údaje o perorálních minimálních a maximálních terapeutických dávkách účinných látek, viz Český lékopis. JKTP 2013
 S AB = 3100 dm 2 I 1 = 8,6 µg/stěr ( plocha stěru = 1 dm 2 ) Údaje o perorálních minimálních a maximálních terapeutických dávkách účinných látek, viz Český lékopis. JKTP")
29
Kriteria přijatelnosti procesní kvalifikace čistícího postupu Limity založené na toxikologických datech Pro látky, u nichž nejsou k dispozici kvantitativní hladiny terapeutické dávky, je výpočet limitů založen na toxicitě materiálu, která je vyjádřena jako LD 50. (např. pro čistící prostředky, pro meziprodukty API, atd.) Pro tyto látky je možné použít i limit 10 ppm jako maximální přenos do šarží zpracovávaných po čištění. Výpočet na základě hodnoty LD 50 lze provést takto: NOEL = LD 50 x EMPIRICKÝ FAKTOR ADI = NOEL x AAW x SF NOEL žádné pozorovaná účinná úroveň Empirický faktor – 0,0005 - odvozen z modelu zkoušky na zvířatech vypracovaného Laytonem ADI přijatelný denní příjem AAW průměrná hmotnost dospělého jedince SF bezpečnostní faktor (pro orální produkty 0,01 – 0,001) I = ADI x NB min. / B max. JKTP 2013
 Pro tyto látky je možné použít i limit 10 ppm jako maximální přenos do šarží zpracovávaných po čištění. Výpočet na základě hodnoty LD 50 lze provést takto: NOEL = LD 50 x EMPIRICKÝ FAKTOR ADI = NOEL x AAW x SF NOEL žádné pozorovaná účinná úroveň Empirický faktor – 0, odvozen z modelu zkoušky na zvířatech vypracovaného Laytonem ADI přijatelný denní příjem AAW průměrná hmotnost dospělého jedince SF bezpečnostní faktor (pro orální produkty 0,01 – 0,001) I = ADI x NB min. / B max. JKTP")
30
Kriteria přijatelnosti procesní kvalifikace čistícího postupu Limity založené na limitech analytické detekce rezidua V některých případech je možné využít pro stanovení limitů reziduí limity analytické detekovatelnosti daných reziduí. Tento postup je vyžadován pro speciální produkty, např. peniciliny, cytostatické látky, silně účinné hormony, apod. Pro některé vysoce účinné látky, zejména ve formě tekutých lékových forem, dosahuje limit získaný výpočtem velmi nízké hodnoty, takže analytická metoda je nedostatečně citlivá. V takovém případě je jakožto kriterium přijatelnosti zvolen limit, který je shodný s mezí detekce analytické metody. JKTP 2013

31
Zásady pro realizaci Recovery testů Pro každou sledovanou látku, u které se vzorky odebírají metodou stěrů, se provádějí Recovery testy. Účelem těchto zkoušek je: - ověřit správný odběr vzorků - ověřit správně zvolené rozpouštědlo - vytvořit podklady pro validaci analytické metody - stanovit Recovery faktor – hodnota, která slouží pro přepočet nalezené koncentrace sledované látky ve stěru Analytické vyhodnocení vzorků - nejčastěji HPLC (metoda musí být validována) JKTP 2013
 JKTP")
32
Vyhodnocení výsledků Na základě výsledků získaných analýzou odebraných vzorků se po korekci na RF provede porovnání této hodnoty a kriteria přijatelnosti. Validace je hodnocena jako vyhovující v případě, kdy je splněno kriterium přijatelnosti – naměřená hodnota je menší nebo rovna požadovanému limitu. V případě nevyhovujícího analytického hodnocení jednoho odebraného vzorku je opakován test daného kritického místa. Zásada: validace je hodnocena jako vyhovující, pokud tři po sobě následující odběry vzorků splňují kriterium přijatelnosti. Pokud během validace jsou dvě a více měření nevyhovujících, je třeba provést rozbor procesu, zjistit příčinu, případně navrhnout nový čistící cyklus. JKTP 2013

33
Analýza posledního oplachového roztoku Kriterium přijatelnosti: kvalitativní parametry vody použité pro závěrečný oplach, tj. vody čištěné, resp. vody pro injekce (dle PhEur) TOC (oxidovatelné látky), dusičnany, těžké kovy, měrná vodivost, mikrobiologická kontrola čistoty Zbytkové množství čistícího prostředku, příp. dalších organických látek (např. pomocných látek) Koncentrace sledované účinné látky JKTP 2013
 TOC (oxidovatelné látky), dusičnany, těžké kovy, měrná vodivost, mikrobiologická kontrola čistoty Zbytkové množství čistícího prostředku, příp. dalších organických látek (např. pomocných látek) Koncentrace sledované účinné látky JKTP")
34
Měření prašnosti, křížové kontaminace a expozičních limitů - tři hodnoty udávající obsah sledované látky v pracovním ovzduší - souvisí s kontaminací produktu vzdušným přenosem - souvisí s bezpečností práce z hlediska toho, na jaké úrovni je personál vystaven účinku sledované látky Princip měření: Odběr vzduchu pomocí čerpadla, kterým je vzorek vzduchu přiváděn na filtr (papírový nebo teflonový). Hmotnostně nebo analyticky je vyhodnocen obsah sledované látky v m 3 vzduchu. JKTP 2013

35
Měření prašnosti, křížové kontaminace a expozičních limitů - měření expozičních limitů – poměrně nový požadavek SVP Kriteria přijatelnosti jsou stanoveny metodami: - na základě hodnoty LD 50 - na základě minimální terapeutické dávky - na základě zavedení tzv. korekčních faktorů JKTP 2013

36
Účinné látky i některé pomocné látky, které se zpracovávají ve farmaceutické výrobě, mohou mít nebezpečné vlastnosti (API-active pharmaceutical ingredient označovány jako „nebezpečné substance“) Vysoce toxické T+, toxické T, dráždivé Xi, karcinogenní k, teratogenní (toxické pro reprodukci r), mutagenní m, zdraví škodlivé Xn Výbušný E, oxidující O, extrémně hořlavý F+, vysoce hořlavý F, žíravý C, nebezpečný pro životní prostředí N Zpracování - Nebezpečí rizika (inhalace, styk s kůží a očima, požití) Nebezpečné vlastnosti látek závisí na toxikologických a farmakologických vlastnostech JKTP 2013
 Vysoce toxické T+, toxické T, dráždivé Xi, karcinogenní k, teratogenní (toxické pro reprodukci r), mutagenní m, zdraví škodlivé Xn Výbušný E, oxidující O, extrémně hořlavý F+, vysoce hořlavý F, žíravý C, nebezpečný pro životní prostředí N Zpracování - Nebezpečí rizika (inhalace, styk s kůží a očima, požití) Nebezpečné vlastnosti látek závisí na toxikologických a farmakologických vlastnostech JKTP 2013")
37
Riziko = nebezpečí x expozice Riziko – možnost poškození vyvolané nebezpečím Nebezpečí – vlastnost předmětu nebo produktu nebo situace – možnost ohrozit zdraví lidí Expozice – kontakt s nebezpečím (lze kontrolovat) JKTP 2013
 JKTP 2013")
38
Riziko = nebezpečí x expozice Je třeba minimalizovat rizika spojená s manipulací s API Technologie výrobního procesu Containment Speciálně navržená výrobní zařízení Speciálně navržené vzduchotechnické ošetření prostoru Standardní Operační Postupy(SOP) OOPP nebo kombinací vyjmenovaných opatření JKTP 2013
 OOPP nebo kombinací vyjmenovaných opatření JKTP 2013")
39
Containment & proč Containment rozumíme ochranu prostoru s přítomností osob (personál firmy, osoby vyskytující se v okolí firmy) před negativním působením (kontaminací) zpracovávaných látek vytvořením „bariery“ * „uzavřená technologie“ výroby přípravku Zabránit expozici personálu s látkami s nebezpečnými vlastnostmi JKTP 2013
 před negativním působením (kontaminací) zpracovávaných látek vytvořením „bariery * „uzavřená technologie výroby přípravku Zabránit expozici personálu s látkami s nebezpečnými vlastnostmi JKTP 2013")
40
Toxikologické vlastnosti: LD 50 - smrtelná dávka pro 50% populace zvířat ve studii (lethal dose for 50% of animal population in study) Farmakologické vlastnosti – účinnost látky (D – dávkování, dose) LD 50 Hormony (tibolon = 2 mg/kg, norethisteron = 2800 mg/kg, ethynilestradiol = 5000 mg/kg) Analgetika (paracetamol = 1944 mg/kg, ibuprofen = 636 mg/kg) D Hormony (tibolon = 2,5 mg/tbl., norethisteron = 5 mg/tbl., ethynilestradiol = 0,02 mg/tbl.) Analgetika (paracetamol = 125 mg/tbl., ibuprofen = 200 mg/tbl.) JKTP 2013
 Farmakologické vlastnosti – účinnost látky (D – dávkování, dose) LD 50 Hormony (tibolon = 2 mg/kg, norethisteron = 2800 mg/kg, ethynilestradiol = 5000 mg/kg) Analgetika (paracetamol = 1944 mg/kg, ibuprofen = 636 mg/kg) D Hormony (tibolon = 2,5 mg/tbl., norethisteron = 5 mg/tbl., ethynilestradiol = 0,02 mg/tbl.) Analgetika (paracetamol = 125 mg/tbl., ibuprofen = 200 mg/tbl.) JKTP 2013")
41
SVP požadavky HSE požadavky Riziko pro pacienta (kvalitativní riziko) Křížová kontaminace Kontaminace obalového materiálu Riziko pro operátora (bezpečnostní riziko) Karcinogenní,teratogenní účinek Alergické reakce JKTP 2013
 Křížová kontaminace Kontaminace obalového materiálu Riziko pro operátora (bezpečnostní riziko) Karcinogenní,teratogenní účinek Alergické reakce JKTP 2013")
42
SVP požadavky HSE požadavky JKTP 2013

43
Farmaceutický výrobce - definovat vlastnosti zpracovávaných látek Důvody: zabránění vzniku křížové kontaminace minimalizace rizik vyplývajících z příp. nebezpečných vlastností zpracovávaných látek předcházení problémům spojených s nemocností personálu a „nemocemi z povolání“ prestiž společnosti, zajištění „dobrého jména firmy“ úspora finančních prostředků JKTP 2013

44
F armaceutické látky (účinné látky, pomocné látky), hodnoty OEL, další doplňující označení, hodnoty OEB OEL – pracovní expoziční limit (occupational exposure limits) Hodnota představuje maximální zdravotně nezávadnou koncentraci účinné látky v ovzduší výrobních prostor [µg / 1 m 3 ], při expozici pracovníka po dobu 8,0 hod. denně po 200 dnů v roce, po dobu 40 let Hodnota uvedená v předpisech, příp. stanovená výpočtem ADI – přijatelný denní příjem (Acceptable Daily Intake) Skutečný příjem látky operátorem za předpokladu úplné absorbce inhalovaného vzduchu JKTP 2013
![F armaceutické látky (účinné látky, pomocné látky), hodnoty OEL, další doplňující označení, hodnoty OEB OEL – pracovní expoziční limit (occupational exposure limits) Hodnota představuje maximální zdravotně nezávadnou koncentraci účinné látky v ovzduší výrobních prostor [µg / 1 m 3 ], při expozici pracovníka po dobu 8,0 hod.](http://images.slideplayer.cz/42/11271389/slides/slide_44.jpg "denně po 200 dnů v roce, po dobu 40 let Hodnota uvedená v předpisech, příp. stanovená výpočtem ADI – přijatelný denní příjem (Acceptable Daily Intake) Skutečný příjem látky operátorem za předpokladu úplné absorbce inhalovaného vzduchu JKTP")
45
Parametry STTD min. Minimální dosažitelná terapeutická denní dávkamg Vobjem vzduchu nadýchaný průměrně za jednu pracovní směnu = 10m 3 /den SFfaktor nejistoty (bezpečnostní) = 1000bezrozm. Např. SF1závažnost účinku = 10 SF2korekce mezi nejnižším pozorovaným účinkem a nepozorovaným účinkem = 10 SF3… JKTP 2013
 = 1000bezrozm. Např. SF1závažnost účinku = 10 SF2korekce mezi nejnižším pozorovaným účinkem a nepozorovaným účinkem = 10 SF3… JKTP")
46
Parametry NOEL žádná pozorovaná účinná úroveň mg/kg x den BWhmotnost personálu (min. 50 kg)kg Vobjem vzduchu nadýchaný průměrně za jednu pracovní směnu = 10 m 3 /den SFfaktor nejistotybezrozm. Empirický faktor odvozen z modelu zkoušky na zvířatech = 0,0005bezrozm. LD 50 smrtelná dávka pro 50% populace zvířat ve studii mg/kg JKTP 2013
kg Vobjem vzduchu nadýchaný průměrně za jednu pracovní směnu = 10 m 3 /den SFfaktor nejistotybezrozm. Empirický faktor odvozen z modelu zkoušky na zvířatech = 0,0005bezrozm. LD 50 smrtelná dávka pro 50% populace zvířat ve studii mg/kg JKTP")
47
OEB – pásmo expozice při práci (occupational exposure band) Souvisí s hodnotou OEL Popisuje podmínky manipulace s danou látkou v souvislosti s jejími nebezpečnými vlastnostmi Pásmo OEB stupnice 1 – 5 Pásmo OEB V1 – V5 pro kapaliny a plyny V různých firmách se liší, např. firma Roche stupnice 1 - 4 JKTP 2013

48
OEB 1: OEL > 1 mg/m3 Může být škodlivá nebo dráždit anebo mít nízkou farmakologickou aktivitu OEB 2: OEL = 0,1 – 1 mg/m3 Škodlivá, může být dráždivá anebo mít střední farmakologickou aktivitu OEB 3: OEL = 10 – 100 µg/m3 Velmi škodlivá, možná toxická nebo může mít vysokou farmakologickou aktivitu OEB 4: OEL = 1,0 – 10 µg/m3 Toxická, velmi toxická, může být korozivní nebo genotoxická anebo mít velmi vysokou farmakologickou aktivitu OEB 5: OEL < 1 µg/m3 Extrémně toxická, může být korozivní nebo genotoxická anebo mít extrémně vysokou farmakologickou aktivitu JKTP 2013

51
EudraLex Volume 4 EU Guidelines for good manufacturing practice for medicinal products for human and veterinary use Velký důraz je kladen na Quality Risk Management proces se zaměřením na toxikologické hodnocení. Na jeho základě pak má výrobce navrhnout příslušná technická a organizační opatření ke zmírnění nebo odstranění rizik souvisejících se vznikem křížové kontaminace a případně navrhnout použití dedikovaných prostor a zařízení. účinnost: března 2015 JKTP 2013

52
EUROPEAN MEDICINES AGENCY Guideline on setting health based exposure limits for use in risk identification in the manufacture of different medicinal products in shared facilities Posouzení křížové kontaminace z hlediska validace čistícího procesu na základě toxikologických dat aktivních substancí účinnost: 1. června 2015 JKTP 2013

53
PDE = Permitted daily Exposure, resp. ADE = Acceptance daily exposure NOAEL = hodnota dávky bez pozorovaného nepříznivého účinku, resp. LOAEL = nejnižší hodnota dávky s pozorovaným nepříznivým účinkem Weight Adjustment = hmotnost jedince [50 kg] F1 – F5 = faktory nejistoty hodnota 1 – 12 (extrapolace mezi druhy, variabilita mezi jedinci, doba trvání, toxikologické vlastnosti (karcinogenita, teratogenita), převod mezi LOAEL a NOAEL) JKTP 2013
, převod mezi LOAEL a NOAEL) JKTP")
54
PASIVACE ZAŘÍZENÍ Na nerezovém povrchu se vlivem působení farmaceutických vod a zpracovávaných látek tvoří oxidy Fe a Cr Na stěru šedé až hnědooranžové Kontaminace, vliv na čistitelnost, nebezpečí vizuální závady produktu Odstranění: působení kyselých mycích prostředků při vysoké teplotě (85°C) po dobu cca 60 min. JKTP 2013

56
ANALÝZA RIZIK Metodika FMEA (Failure Mode and Effect Analysis) Analýza chybových stavů Hodnotí se Závažnost, Četnost a Detekovatelnost chyby V případě střední a vysoké míry rizika je třeba přijmout nápravná opatření Opakované vyhodnocení Četnosti a Detekovatelnosti s cílem ověřit, že v důsledku přijatých nápravných opatření je riziko přijatelné JKTP 2013
 Analýza chybových stavů Hodnotí se Závažnost, Četnost a Detekovatelnost chyby V případě střední a vysoké míry rizika je třeba přijmout nápravná opatření Opakované vyhodnocení Četnosti a Detekovatelnosti s cílem ověřit, že v důsledku přijatých nápravných opatření je riziko přijatelné JKTP 2013")
57
ANALÝZA RIZIK Metodika SANOFI, QOQS – 003227 Spočívá v posouzení hodnocených kritérií (parametrů) pro výrobu přípravku a nebezpečnosti (kritičnosti) přípravku Nebezpečnost (kritičnost) je vyjádřena hodnotou OEL (pracovní expoziční limit), resp. OEB (pracovní expoziční pásmo) Míra rizika je odečtena z matice rizika, v níž vertikální osa odpovídá výskytu daného kriteria a vodorovná osa kritičnosti hodnoceného přípravku. JKTP 2013
 Míra rizika je odečtena z matice rizika, v níž vertikální osa odpovídá výskytu daného kriteria a vodorovná osa kritičnosti hodnoceného přípravku. JKTP")
58
ANALÝZA RIZIK Metodika SANOFI, QOQS – 003227 OEB 1,2 – nízký, OEB 3 – střední, OEB 4 – vysoký, OEB 5 – velmi vysoký JKTP 2013 NepřijatelnýNejistýPřijatelný Nepřijatelný stav Nutná nápravná opatření Nejistý stav Nutná kontrolní měření Jistý (jasný) stav Monitoring
 stav Monitoring")
59
NOVINKY Použití černých utěrek pro účely zdokonalení vizuální kontroly čistoty Snížení limitu ze 400 µg/stěr až na 50 µg/stěr JKTP 2013

60
Chemická dekontaminace reziduí účinných látek Přípravek Endiex kapsle, účinná látka Nifuroxazid Obtížně čistitelné z důvodu barevnosti a nerozpustnosti v čistícím mediu Použití dezinfekčního přípravku (Deconex Surface AF) Dochází k hydrolýze účinné látky, která zvýší iontový charakter látky a tedy i rozpustnost (žlutá barva látky přechází na tmavě červenou) JKTP 2013
 Dochází k hydrolýze účinné látky, která zvýší iontový charakter látky a tedy i rozpustnost (žlutá barva látky přechází na tmavě červenou) JKTP 2013")
61
Použití přístroje ITMS (ion trap mobility spektrometry) Patentovaná technologie původně vyvinutá pro bezpečnostní aplikaci (letiště…) Identifikace látek se provádí na základě srovnání doby průletu ionizované molekuly analytu a standardu API urychlovací trubicí Přímý stěr pomocí „polymidových zlatých trapů“ + velmi rychlá analýza, selektivní a citlivé, minimální spotřeba chemikálií - dlouhý vývoj metody, vhodné pro API o určitém bodu tání a molekulové hmotnosti, pořizovací cena JKTP 2013
 Patentovaná technologie původně vyvinutá pro bezpečnostní aplikaci (letiště…) Identifikace látek se provádí na základě srovnání doby průletu ionizované molekuly analytu a standardu API urychlovací trubicí Přímý stěr pomocí „polymidových zlatých trapů + velmi rychlá analýza, selektivní a citlivé, minimální spotřeba chemikálií - dlouhý vývoj metody, vhodné pro API o určitém bodu tání a molekulové hmotnosti, pořizovací cena JKTP 2013")
63
KVÍZ Pojmem „křížová kontaminace“ rozumíme: A) Operátora znečištěného použitím WC B) Hotové balení přípravku, v němž se nachází jedna nebo více tablet jiného přípravku C) Produkt znečištěný pomocnou látkou, která není ve složení Jako kriterium přijatelnosti čistoty výrobního zařízení po ukončení výroby přípravku ze skupiny cytostatik je zvoleno: A) Senzorické hodnocení povrchu zařízení (chuťové, čichové posouzení) B) Vizuální posouzení čistoty C) Limit dostupné analytické metody JKTP 2013
 Operátora znečištěného použitím WC B) Hotové balení přípravku, v němž se nachází jedna nebo více tablet jiného přípravku C) Produkt znečištěný pomocnou látkou, která není ve složení Jako kriterium přijatelnosti čistoty výrobního zařízení po ukončení výroby přípravku ze skupiny cytostatik je zvoleno: A) Senzorické hodnocení povrchu zařízení (chuťové, čichové posouzení) B) Vizuální posouzení čistoty C) Limit dostupné analytické metody JKTP 2013")
64
V přípravku obsahujícím účinnou látku Paracetamol je zjištěna kontaminace návykovou látkou Pseudoephedrin hydrochlorid v množství 0,1 mg/tbl. ÚKOL: Je kontaminace paracetamolových tablet v limitu ? Posoudit, zda-li muž užívající velkou dávku tohoto analgetika bude vyžadovat večer na své partnerce určité aktivity z důvodu dostavující se nespavosti JKTP 2013

65
1 tableta Paracetamolu obsahuje 500 mg účinné látky Maximální terapeutická dávka Paracetamolu : 4 g /den, tzn. 8 tablet V maximální dávce pacient spolyká 0,8 mg Pseudoephedrinu hydrochloridu Minimální terapeutická dávka Pseudoephedrinu HCl: 30 mg Použijeme limit I 1 (str. 21) 1/1000 Pseudoephedrinu HCl: 0,03 mg 0,8 > 0,03 JKTP 2013
 1/1000 Pseudoephedrinu HCl: 0,03 mg 0,8 > 0,03 JKTP")
66
DĚKUJI ZA POZORNOST JKTP 2013

Podobné prezentace