Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Molekulová a kvantová mechanika

2
Opakování z minula Hierarchie teoretických metod –počítačová chemie – simulace na atomární úrovni ab initio (QM) MM/MD –Ostatní metody QSAR, bioinformatika, systémová biologie
 MM/MD –Ostatní metody QSAR, bioinformatika, systémová biologie")
3
Molekulová dynamika počítačová simulace atomy/molekuly interagují po jistou dobu dle zákonů KLASICKÉ fyziky výsledkem je pohled na časový vývoj systému interaguje obecně mnoho částic a není možno zjistit dynamické vlastnosti takového systému analyticky => numerika

4
Proč, když máme QM? běžná nepoužitelná malá molekula kvantový chemik jásá střední molekula - kvantovému chemikovi tuhnou rysy a volí velmi umírněné prostředky běžný protein – kvantový chemik pláče a s hanbou prchá z boje

5
Born-Oppenheimerova aproximace oddělení elektronického a jaderného pohybu kvantové elektrony vs. klasická jádra celkovou energii systému je možno psát jako funkci pozice jader E = f(R) a popsat aparátem klasické fyziky
 a popsat aparátem klasické fyziky.")
6
PES (Potential Energy Surface) přímý důsledek Born-Oppenheimerovy aproximace energie molekuly v základním stavu je funkcí toliko souřadnic jejích jader –při změně polohy jader se mění energie molekuly –změna polohy – např. rotace kolem vazby energetická cena závisí na typu změny –změna C-C o 0.1 Å, cca 3 kcal.mol -1 –změna Ar... Ar o 1 Å, cca 0.1 kcal.mol -1

7
A. R. Leach, Molecular Modelling, 2001 v tomto případě je PES fcí pouze jedné souřadnice (torze) stacionární body – první derivace energie je 0, síly na atomy jsou 0 minima na PES odpovídají stabilním strukturám a jsou jedním z možných stacionárních bodů
 stacionární body – první derivace energie je 0, síly na atomy jsou 0 minima na PES odpovídají stabilním strukturám a jsou jedním z možných stacionárních bodů.")
8
čili my potřebujeme nějak vyjádřit energii systému jako funkci souřadnic jader to je doménou větve počítačové chemie nazývající se molekulová mechanika (či metody silového pole – force field) tyto metody tedy zanedbávají elektronický pohyb a tudíž je není možno použít na popis vlastností/jevů na elektronech závisejících (např. vznik/zánik vazeb)
.")
9
Empirický potenciál energie jako funkce pozice atomů (jader) je konstruována jako empirický potenciál (silové pole) to znamená, že celkovou energii molekuly rozbijeme na menší části, ty nějak vypočítáme a pak to všechno posčítáme dohromady molekulová mechanika MM
 je konstruována jako empirický potenciál (silové pole) to znamená, že celkovou energii molekuly rozbijeme na menší části, ty nějak vypočítáme a pak to všechno posčítáme dohromady molekulová mechanika MM")
10
Empirický potenciál

11
molekulová mechanika je založena na dosti jednoduchém modelu interakcí v rámci systému s příspěvky z procesů jako je bond stretching, angle bending, rotation around bond aditivita síla každého z těchto příspěvků je popsána parametry, které jsou nějak určeny (parametrizace) – empirická metoda transferabilita
 – empirická metoda transferabilita")
12
vazebné příspěvkynevazebné příspěvky

13
parametry

14
celková energie systému je popsána jako součet energetických penalt spojených s deformacemi vazeb, úhlů a torzí (rotace) z jejich referenčních („rovnovážných“) poloh + příspěvky popisující interakce mezi částmi molekuly jež nejsou kovalentně vázány
 z jejich referenčních („rovnovážných ) poloh + příspěvky popisující interakce mezi částmi molekuly jež nejsou kovalentně vázány")
15
silové pole (ff) – nejen funkční tvar termů, ale i parametry v nich vystupující ff jsou primárně postaveny tak, aby reprodukovaly strukturní vlastnosti ff je třeba použít na ty vlastnosti, na které byly parametrizovány (např. strukturní na struktury, na spektra jiné) transferabilita fční formy i parametrů je důležitá, leč někdy je dobré vytvořit parametry pouze pro určitou molekulu
 transferabilita fční formy i parametrů je důležitá, leč někdy je dobré vytvořit parametry pouze pro určitou molekulu.")
16
ff jsou empirické -> neexistuje „správný“ ff funkční formy jsou kompromisem mezi přesností a výpočetní náročností + pro minimalizaci či MD je třeba počítat první a druhé derivace energie dle souřadnic NÁZVOSLOVÍ energie – empirický potenciál silové pole (force field) – funkční tvar příspěvků i sada parametrů pro jednotlivé příspěvky
 – funkční tvar příspěvků i sada parametrů pro jednotlivé příspěvky")
17
Atomový typ QM MM –každému atomu je třeba přiřadit atomový typ –ten nese informaci nejen o tom o jaký atom se jedná, ale i o hybridizaci a někdy o okolí uhlík sp 3 (referenční úhel je 109.5˚), sp 2 (120˚) uhlík v MM2/3/4 - sp 3, sp 2, sp, carbonyl, cyclopropane, radical, cyclopropene, carbonium atomová čísla, geometrie, náboj, spin
, sp 2 (120˚) uhlík v MM2/3/4 - sp 3, sp 2, sp, carbonyl, cyclopropane, radical, cyclopropene, carbonium atomová čísla, geometrie, náboj, spin")
18
C -CA-CA 63.0 120.00 CA-CA-CA 63.0 120.00 CA-CA-CB 63.0 120.00 CA-CA-CT 70.0 120.00 CA-CA-HA 35.0 120.00 X-CT-CT-X91.400.03. CT-CT-OS-CT10.3830.-3. CT-CT-OS-CT10.1180.02.

19
Nejpoužívanější ff malé organické molekuly –Allinger et al. – MM2, MM3, MM4 http://europa.chem.uga.edu/allinger/mm2mm3.html biomolekuly –Amber ff (ff94, ff96, ff98, ff99) – jak proteiny, tak NA, lepší jsou pro NA http://amber.scripps.edu/ –CHARMM (19, 22, 27) - jak proteiny, tak NA, lepší jsou pro proteiny http://www.charmm.org/
 – jak proteiny, tak NA, lepší jsou pro NA –CHARMM (19, 22, 27) - jak proteiny, tak NA, lepší jsou pro proteiny")
20
Volně dostupné programy Tinker –http://dasher.wustl.edu/tinker/http://dasher.wustl.edu/tinker/ –ff94,96,98,99; CHARMM 19,27; MM2, MM3, OPLS, polarizovatelný ff Amoeba Gromacs –http://www.gromacs.org/http://www.gromacs.org/ –Gromos, OPLS, Amber ff (http://chemistry.csulb.edu/ffamber/)http://chemistry.csulb.edu/ffamber/

21
Silové pole I

22
vazebné příspěvkynevazebné příspěvky

24
Deformace vazebné vzdálenosti zajímá nás chování kolem minima

25
v MM je vzácností že by se vazby výrazně odlišovaly od rovnovážné délky v okolí rovnovážné délky je možno potenciál popsat Hookovým zákonem k je silová konstanta, r 0 je referenční délka vazby parametry: r 0, k

26
Hookův zákon

27
Různé vazby = různé pružiny dva parametry: r 0 k Molekulakr0r0 H251074,1 HCl478127,5 HBr408141,4 HI291160,9

28
síly mezi vázanými atomy jsou značné, je potřeba hodně energie na vychýlení, silové konstanty k jsou velké silnější vazby maji k vyšší (C-C vs. C=C) A. R. Leach, Molecular Modelling, 2001
 A. R. Leach, Molecular Modelling,")
29
Parametry jak ale získám parametry r 0 a k? experimenty –geometrie: X-ray, NMR, rotační spektroskopie –silové konstanty: vibrační spektroskopie výpočtem –QM vypočtu povrch potenciální energie a potom analyticky nafituji na tyto body křivku

30
pro popis širokého rozsahu deformací vazeb se používá Morseho potenciál D e je hloubka minima, a = ω sqrt(μ/2D e ) kde μ je redukovaná hmotnost m 1 m 2 /(m 1 +m 2 ), ω je frekvence vibrace vazby, l 0 je referenční délky vazby parametry: D e, ω, l 0
 kde μ je redukovaná hmotnost m 1 m 2 /(m 1 +m 2 ), ω je frekvence vibrace vazby, l 0 je referenční délky vazby parametry: D e, ω, l 0")
31
A. R. Leach, Molecular Modelling, 2001 Hookův zákon

32
Změna velikosti úhlu Hookův zákon

33
Torzní členy natahování vazeb a ohýbání úhlů – „hard“ degrees of freedom (je třeba hodně energie na vyvolání deformace z jejich referenční hodnoty) většina variace ve struktuře a relativních energiích je způsobena komplexní souhrou mezi torzními a nevazebnými příspěvky
 většina variace ve struktuře a relativních energiích je způsobena komplexní souhrou mezi torzními a nevazebnými příspěvky")
34
A. R. Leach, Molecular Modelling, 2001 torzní člený popisují bariéry rotace kolem chemických vazeb najdou se všechny vázané „kvartety“ (9 v ethanu), každý z nich je popsán nějakým torzním potenciálem
, každý z nich je popsán nějakým torzním potenciálem.")
35
torzní potenciál se téměř výhradně vyjadřuje jako kosinovská série parametry: V n – výška bariéry, n – multiplicita (počet minim), γ - fáze
, γ - fáze")
36
etan (rotace kolem dvou sp 3 uhlíků): n = 3, γ = 0˚
: n = 3, γ = 0˚")
37
A. R. Leach, Molecular Modelling, 2001

39
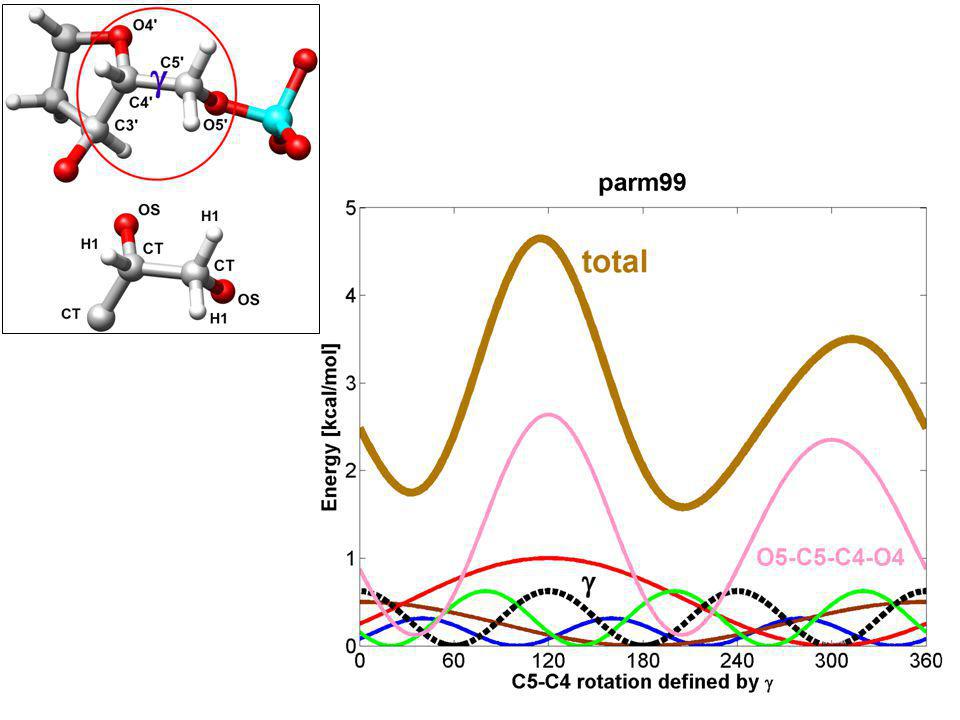
Amber ff –mnoho torzních příspěvků obsahuje pouze jeden člen v expanzi –ale např. pro správný popis preference gauche konformace O-C-C-O vazby (OCH 2 - CH 2 O fragment v cukru DNA) je potřeba dvou cos v torzním potenciálu
 je potřeba dvou cos v torzním potenciálu.")
Podobné prezentace
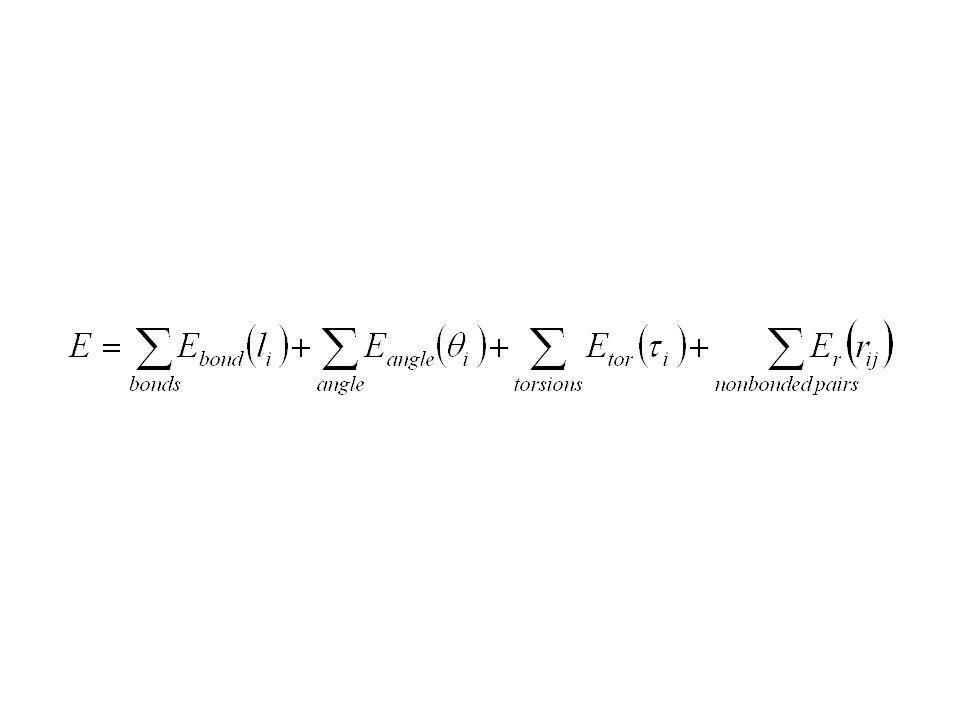














 –klasická fyzika PES (Potential.>")

 REZONANCE>")

 1>")

