Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
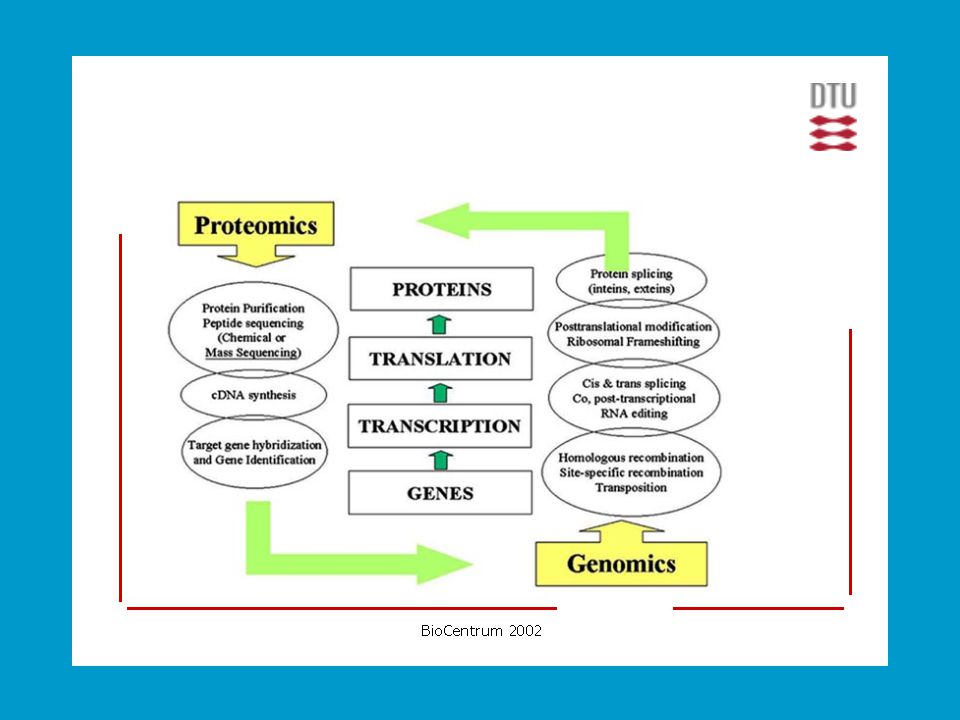
Definování proteomu Znalost úplné genetické informace organismu (genomu), získaná sekvencováním DNA a určením funkce jednotlivých genů umožňuje hlouběji proniknout do molekulové podstaty funkce živých organismů. Cílem rozluštit genom se zabývá vědní odvětví zvané genomika. Genetická informace ale hovoří pouze o dědičně převzatých schopnostech organismu. K realizaci životních funkcí organismu je nutné na podkladu genů vytvářet proteiny. Tento děj je podléhá regulaci zavisející na mnoha vnitřních i vnějších faktorech. Pro soubor všech buněčných proteinů se od 1995 (Wilkins) ujal pojem proteom jako komplementární soubor proteinů k souboru genů (genomu). PROTeins expressed by a genOME.
, získaná sekvencováním DNA a určením funkce jednotlivých genů umožňuje hlouběji proniknout do molekulové podstaty funkce živých organismů. Cílem rozluštit genom se zabývá vědní odvětví zvané genomika. Genetická informace ale hovoří pouze o dědičně převzatých schopnostech organismu. K realizaci životních funkcí organismu je nutné na podkladu genů vytvářet proteiny. Tento děj je podléhá regulaci zavisející na mnoha vnitřních i vnějších faktorech. Pro soubor všech buněčných proteinů se od 1995 (Wilkins) ujal pojem proteom jako komplementární soubor proteinů k souboru genů (genomu). PROTeins expressed by a genOME.")
2
Pojem proteom vystihuje souhrn všech proteinů, které jsou kódované a exprimované v buňce (či viru) spolu s jejich interakcí (proteinové komplexy) a funkčními vztahy (tzv. specifické souvislosti). Jiná definice říká, že jde o okamžitý soubor všech proteinů ve vzorku (buňka, tkáň) - tedy v určitém čase. Obdobně třeba lipidom, lipidomika.
 - tedy v určitém čase. Obdobně třeba lipidom, lipidomika.")
3
Lokalizace a funkce proteinových komplexů (PK)
")
4
Proteinové komplexy a jejich hyperspojení
nebo Jádro proteomu (konstelace stabilních PK a volných proteinů) nad to: přesahové interakce, regulátorové proteiny, buněčně specifické modifikace
 nad to: přesahové interakce, regulátorové proteiny, buněčně specifické modifikace.")
5
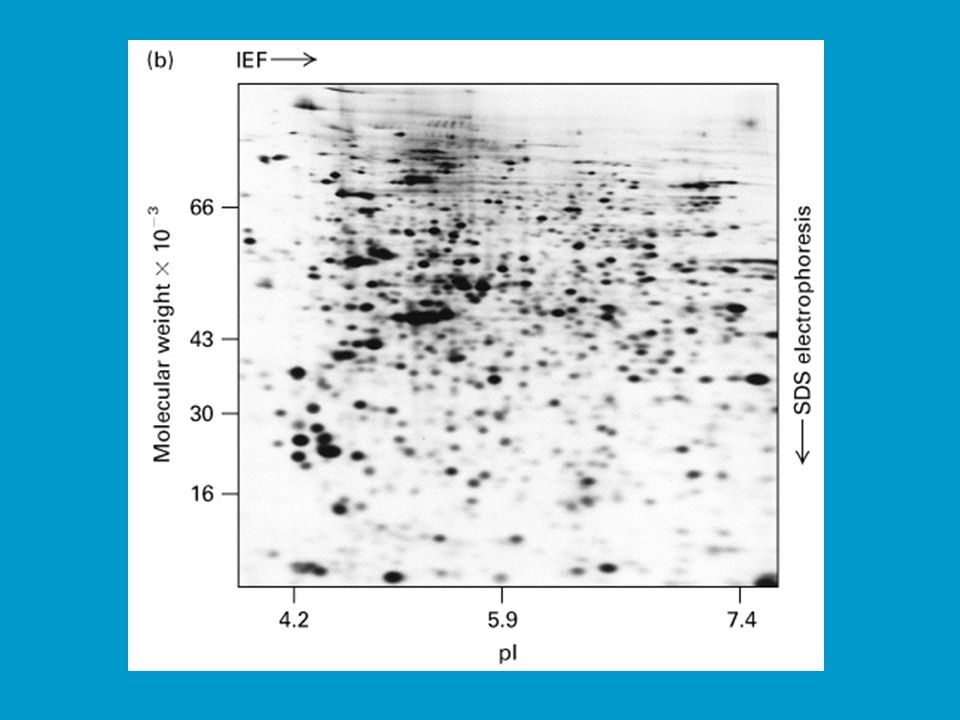
Proteomika Zatímco genom je pojmem jednoznačně statickým, v podstatě konstantním, proteom je pojmem dynamickým. Koncentrace proteinů se totiž neustále mění podle aktuálních potřeb organismu (na rozdíl od genové výbavy). Studiem proteomu se zabývá vědní odvětví na rozhraní Bi-Ch, proteomika. Cílem je komplexní analýza proteinů organely, buňky, tkáně či extracelulárních tekutin. Na počátku takové analýzy vždy stojí separace složité směsi proteinů v extraktech elektro-foretickými metodami. V tomto směru největší potenciál má 2-D elektroforéza (ale i obě dimenze jednotlivě, dle potřeby). Získá se takto tzv. proteinová mapa. Proteiny z proteinové mapy jsou pak identifikovány na základě obecného postupu 1. Edmanova sekvenční analýza nebo MS, 2. vyhledávání v dtb.
. Studiem proteomu se zabývá vědní odvětví na rozhraní Bi-Ch, proteomika. Cílem je komplexní analýza proteinů organely, buňky, tkáně či extracelulárních tekutin. Na počátku takové analýzy vždy stojí separace složité směsi proteinů v extraktech elektro-foretickými metodami. V tomto směru největší potenciál má 2-D elektroforéza (ale i obě dimenze jednotlivě, dle potřeby). Získá se takto tzv. proteinová mapa. Proteiny z proteinové mapy jsou pak identifikovány na základě obecného postupu 1. Edmanova sekvenční analýza nebo MS, 2. vyhledávání v dtb.")
7
CIBULOVÝ MODEL BIOLOGICKÉHO VÝZKUMU

8
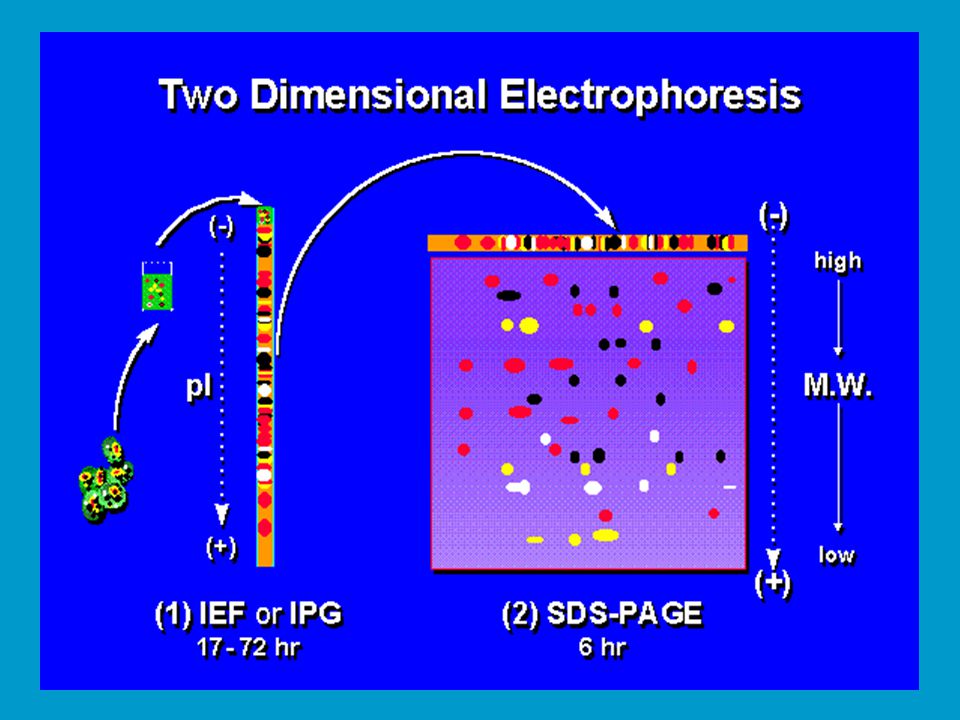
2D elektroforéza Kombinuje použití IEF a SDS-PAGE pro účely studia proteomu. vypracováno O´Farrellem v 70. letech, dnes do značné míry polo- automatizovaná rutinní záležitost. Dodnes nepřekonaná komplexní elektroforetická separace proteinů. Směs proteinů nejprve separována IEF v přítomnosti močoviny, a to buď v trubičkovém gelu nebo na tzv. stripu plochého gelu. Následně je IEF gel přenesen na plochý gel pro SDS-PAGE a elektroforéza proběhne ve směru 2. dimenze (90o vůči 1. dimenzi). Dnes se používá komerčních gelů pro obě dimenze, dosti drahá záležitost. Rutinně lze takto separovat proteinů v závislosti na formátu gelu.
. Dnes se používá komerčních gelů pro obě dimenze, dosti drahá záležitost. Rutinně lze takto separovat proteinů v závislosti na formátu gelu.")
10
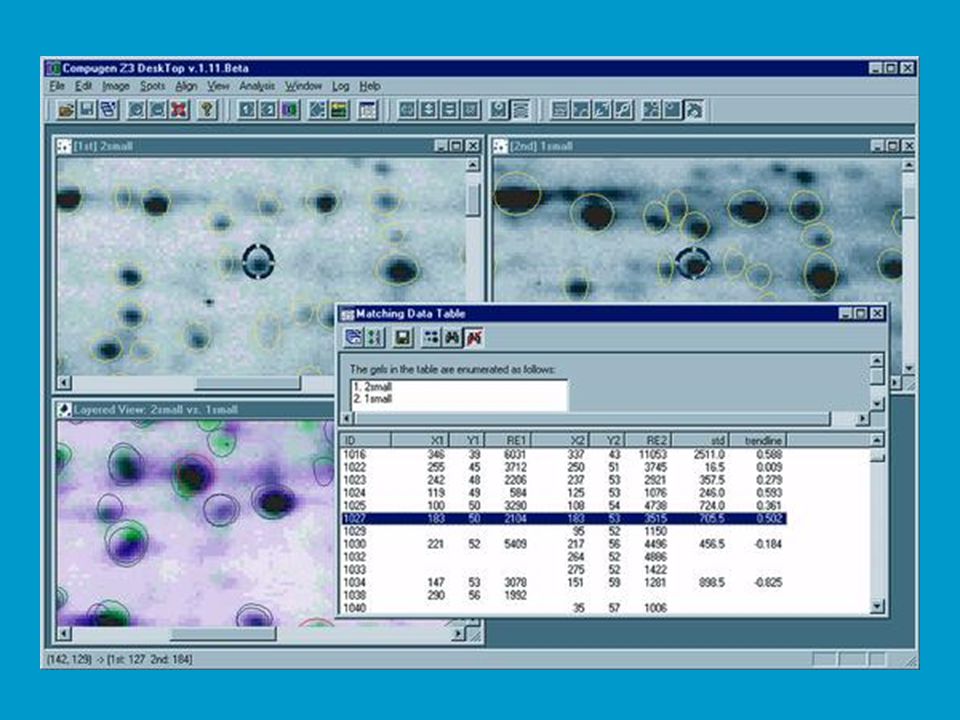
2D elektroforéza je důležitá pro analýzu proteinů v totálních buněčných extraktech. Na 2D elektroforéze a následné sekvenční analýze proteinů je postavena proteomika jako jedno z rozvíjejících se odvětví life sciences. Jak bylo řečeno proteom vystihuje souhrn všech proteinů, které jsou kódované a exprimované v buňce (či viru) spolu s jejich interakcí a funkčními vztahy (tzv. specifické souvislosti). Analýza proteomu začíná separací proteinů v extraktech 2D elektroforézou; následuje počítačová „image analysis“ gelu, při které se například ověřují změny v poloze tzv. spotů příslušejících určitým proteinům, ke kterým dochází na základě přirozených nebo uměle vyvolaných změn v buňce. Lze tak například porovnávat standardní organismy („wild type“) s mutanty, vyšlechtěné odrůdy, hodnotit vliv stresu.
 spolu s jejich interakcí a funkčními vztahy (tzv. specifické souvislosti). Analýza proteomu začíná separací proteinů v extraktech 2D elektroforézou; následuje počítačová „image analysis gelu, při které se například ověřují změny v poloze tzv. spotů příslušejících určitým proteinům, ke kterým dochází na základě přirozených nebo uměle vyvolaných změn v buňce. Lze tak například porovnávat standardní organismy („wild type ) s mutanty, vyšlechtěné odrůdy, hodnotit vliv stresu.")
13
Identifikace proteinů v proteomice
Molekulovou hmotnost nativního proteinu separovaného ELFO lze potvrdit hmotovou spektroskopií MALDI TOF, ESI TOF. Sekvenční analýza se provádí způsoby: 1.Edmanovou degradací (přímé sekvencování) a 2. hmotovou spektrometrií (nepřímé sekvencování, fingerprinting, „de novo“ sekvencování). V obou případech pak na základě získaných sekvencí následuje hledání v databázích proteinů Pro přímé sekvencování je nutný blotting nativního proteinu nebo jeho peptidových fragmentů (připravených in-solution štěpením a následně separovaných ELFO Schäger/von Jagow) na PVDF membránu. Po obarvení membrány amidočerní 10B se vyříznou příslušné pásy (1D) nebo skvrny (2D) a fragmenty membrány se vloží do reakční cely proteinového sekvenátoru.
 a 2. hmotovou spektrometrií (nepřímé sekvencování, fingerprinting, „de novo sekvencování). V obou případech pak na základě získaných sekvencí následuje hledání v databázích proteinů. Pro přímé sekvencování je nutný blotting nativního proteinu nebo jeho peptidových fragmentů (připravených in-solution štěpením a následně separovaných ELFO Schäger/von Jagow) na PVDF membránu. Po obarvení membrány amidočerní 10B se vyříznou příslušné pásy (1D) nebo skvrny (2D) a fragmenty membrány se vloží do reakční cely proteinového sekvenátoru.")
14
coupling cleavage conversion

15
Detailně průběh 3 x C

16
Auromatizace

17
Po odštěpení PTC-derivátu následuje jeho analýza (zjištění retenčního času na HPLC koloně) s detekcí při 270 nm. Běžně je takto možné získat aminokyselin z N-konce, samozřejmě s narůstající chybou (reakce není 100% ní). Za ideálních posmínek maximálně 100 aminokyselin. V automatickém proteinovém sekvenátoru (dodávají např. firmy Applied Biosystems, Shimadzu) je analyzovaný protein či peptid imobilizován. Buď na disku ze skleněných vláken (kovalentní vazba nebo adsorpce) nebo na membráně PVDF (ProSorb patrony nebo po blottingu). Reagencie ke vzorku vstupují a jsou odváděny nejlépe v plynné fázi. Doba trvání jednoho cyklu analýzy činí min. Je třeba připočítat eluci standardů aminokyselin a tzv. prázdný run.
. Za ideálních posmínek maximálně 100 aminokyselin. V automatickém proteinovém sekvenátoru (dodávají např. firmy Applied Biosystems, Shimadzu) je analyzovaný protein či peptid imobilizován. Buď na disku ze skleněných vláken (kovalentní vazba nebo adsorpce) nebo na membráně PVDF (ProSorb patrony nebo po blottingu). Reagencie ke vzorku vstupují a jsou odváděny nejlépe v plynné fázi. Doba trvání jednoho cyklu analýzy činí min. Je třeba připočítat eluci standardů aminokyselin a tzv. prázdný run.")
18
Pro analýzu Edmanovou degradací jsou nutná množství vzorku pmol, nejlépe však 200 pmol, konkurenční MS nyní zvládá fmoly (nanosprej ESI Qq TOF MS), dosud však není tolik rozšířená. Podmínkou pro úspěšnou sekvenční analýzu Edmanovou cestou je volný N-konec peptidu či proteinu. Často je však N-konec chemicky blokován (acetylace, pyroglutamová kyselina). Nevýhodou i výskyt glykosylačních míst - zde nízký výtěžek v daném cyklu. V praxi se blokace dá obejít analýzou vnitřních (interních) peptidů (viz dále), nebo chemickou či enzymovou (pyro-glutaminpeptidasa) deblokací a enzymovou deglykosylací.
. Nevýhodou i výskyt glykosylačních míst - zde nízký výtěžek v daném cyklu. V praxi se blokace dá obejít analýzou vnitřních (interních) peptidů (viz dále), nebo chemickou či enzymovou (pyro-glutaminpeptidasa) deblokací a enzymovou deglykosylací.")
20
Hmotnostní (hmotová) spektrometrie mass spectometry, MS
Je metoda pro separaci látek podle rozdílů hmoty (m) a náboje (z) s využitím elektrického a magnetického pole. Určovanou fyzikální veličinou je podíl hmoty a náboje (m/z), při znalosti náboje umožňuje určit molekulovou hmotnost. Velmi citlivá metoda, stačí několik iontů k získání měřitelného signálu, poměr m/z může být stanoven s přesností 10-4. Původně aplikována pro malé těkavé látky, či derivatizované netěkavé látky (ionizace paprskem elektronů - EI, chemická ionizace s nosným plynem). S objevem šetrnějších technik ionizace (ESI, MALDI) je možné měřit i velké molekuly (např. proteiny, lipidové komplexy, polysacharidy).
 a náboje (z) s využitím elektrického a magnetického pole. Určovanou fyzikální veličinou je podíl hmoty a náboje (m/z), při znalosti náboje umožňuje určit molekulovou hmotnost. Velmi citlivá metoda, stačí několik iontů k získání měřitelného signálu, poměr m/z může být stanoven s přesností Původně aplikována pro malé těkavé látky, či derivatizované netěkavé látky (ionizace paprskem elektronů - EI, chemická ionizace s nosným plynem). S objevem šetrnějších technik ionizace (ESI, MALDI) je možné měřit i velké molekuly (např. proteiny, lipidové komplexy, polysacharidy).")
21
Biochemické aplikace hmotové spektrometrie
Velký rozvoj přístrojů a metod pro MS v poslední dekádě dramaticky zvýšil uplatnění metody pro výzkum v biochemii. Jde o studium biomakromolekul a jejich konglomerátů zaměřený převážně na proteiny. LC-MS se používá pro peptidové mapování, studium disulfidových vazeb v proteinech (srovnání mapy intaktního a redukovaného proteinu). MS se stala jedním z článků studia proteomu. Proteomová analýza (nejčastěji s MALDI TOF MS) často navazuje na laboratorní studium exprese genů, zvláště při genových manipulacích: identifikace rekombinantních proteinů, posouzení mikroheterogenity, posttranslační modifikace.
. MS se stala jedním z článků studia proteomu. Proteomová analýza (nejčastěji s MALDI TOF MS) často navazuje na laboratorní studium exprese genů, zvláště při genových manipulacích: identifikace rekombinantních proteinů, posouzení mikroheterogenity, posttranslační modifikace.")
22
Electrospray ionisation (ESI)
Při této metodě je vzorek rozpuštěn v těkavém rozpouštědle a rozprašován pomocí nabité mikrostříkačky (kovová, skleněná s kov. pístem - Hamilton). Vzniká tak aerosol drobných kapiček, který je vysoušen proudem suchého dusíku. Jak klesá velikost kapičky, roste hustota náboje. Dojde k tzv. kulombické explozi, při které se uvolní ionty, které odchází do spektrometru. Podobně jako u techniky MALDI (viz dále), vzorek je disintegrován na jednotlivé molekuly a ionizován při těch nejjemnějších podmínkách. V ideálním případě by vzorek měl být rozpuštěn v čistém rozpouštědle, u reálných vzorků biomakromolekul je někdy nutné zachovat pufry kvůli stabilitě, musí ale být velmi zředěné. U ESI je velmi výhodné použití např. uhličitanu amonného, HAc, HCOOH. Ionty z jiných pufrů mohou ve spektru interferovat se studovanou látkou.
. Vzniká tak aerosol drobných kapiček, který je vysoušen proudem suchého dusíku. Jak klesá velikost kapičky, roste hustota náboje. Dojde k tzv. kulombické explozi, při které se uvolní ionty, které odchází do spektrometru. Podobně jako u techniky MALDI (viz dále), vzorek je disintegrován na jednotlivé molekuly a ionizován při těch nejjemnějších podmínkách. V ideálním případě by vzorek měl být rozpuštěn v čistém rozpouštědle, u reálných vzorků biomakromolekul je někdy nutné zachovat pufry kvůli stabilitě, musí ale být velmi zředěné. U ESI je velmi výhodné použití např. uhličitanu amonného, HAc, HCOOH. Ionty z jiných pufrů mohou ve spektru interferovat se studovanou látkou.")
23
Electrospray ionisation (ESI)
v proteomice uspořádání „nanospray“ - malé objemy vzorku

24
Matrix-assisted laser desorption ionization (MALDI)
Laserová desorpční ionizace s pomocí matrice Metoda MALDI MS (od 1988) je rozšířeným nástrojem pro separaci přírodních látek, peptidů, proteinů a dalších bio-makromolekul (oligonukleotidy, sacharidy, lipidy). Vzorek je na nerezové destičce ukotven v netěkavé matrici (kokrystalizace). Používá se např. kyselina nikotinová nebo 2,5-dihydroxybenzoová. Vzorek (1 mg/mL) se nanese v množství 0.5 ml na nerezovou destičku a nechá se vysušit. Pak se aplikuje 0.5 ml matrice a opět se nechá vysušit. Matrice se volí dle vzorku, účelem použití je desorpce energie laseru. Destička se vloží do přístroje, zacílí se laserový paprsek („fire“), transfer energie způsobí ionizaci. Směrovaný energetický impuls poskytuje vysoké výtěžky iontů intaktního analytu, dosahuje se subpikomolární sensitivity.
 je rozšířeným nástrojem pro separaci přírodních látek, peptidů, proteinů a dalších bio-makromolekul (oligonukleotidy, sacharidy, lipidy). Vzorek je na nerezové destičce ukotven v netěkavé matrici (kokrystalizace). Používá se např. kyselina nikotinová nebo 2,5-dihydroxybenzoová. Vzorek (1 mg/mL) se nanese v množství 0.5 ml na nerezovou destičku a nechá se vysušit. Pak se aplikuje 0.5 ml matrice a opět se nechá vysušit. Matrice se volí dle vzorku, účelem použití je desorpce energie laseru. Destička se vloží do přístroje, zacílí se laserový paprsek („fire ), transfer energie způsobí ionizaci. Směrovaný energetický impuls poskytuje vysoké výtěžky iontů intaktního analytu, dosahuje se subpikomolární sensitivity.")
25
MALDI

26
DE (delayed extraction) - ionty se okamžitě po ionizaci ochladí (~150 ns), tím se sníží distribuce kinetické energie (ionty se stejnou hodnotou m/z mají různou kinetickou energii). Použití pro látky pod 30 kDa.

27
Některé používané matrice
Dodáním energie dojde k odpaření matrice, která se nachází v nadbytku; v plynné fázi pak matrice nese analyt. Analyt je tak převeden do plynné fáze nepřímo. Matrice je zároveň donorem či akceptorem protonu, podle modu ionizace. Vůbec první byla kyselina nikotinová.

28
Schéma hmotového spektrometru

29
MS je často propojena s jinými analytickými technikami, ať už separačními nebo bez separace. Vznikají tak systémy GC-MS, LC-MS, CE-MS ale i MS-MS. Kromě tandemové MS (MS-MS) mohou být spojeny až tři i více analyzátorů („multiple“ MS). První vybírá studované látky ze směsi, druhý je fragmentuje, třetí analyzuje vzniklé fragmenty. Pro identifikaci látek ve složitých směsích; nikoli na základě m/z, ale podle fragmentačních obrazů („patterns“). Ve spojení s chromato či elfo metodou slouží MS jako detektor s vysokým rozlišením. Taková on-line analýza pak dává okamžité výsledky na rozdíl od off-line detekce. Klíčové je propojení přístrojů. LC-MS v biochemii pro peptidy a proteiny, GC-MS pro těkavé LMW látky, CE-MS široký záběr od LMW po proteiny, DNA, viry etc.
 mohou být spojeny až tři i více analyzátorů („multiple MS). První vybírá studované látky ze směsi, druhý je fragmentuje, třetí analyzuje vzniklé fragmenty. Pro identifikaci látek ve složitých směsích; nikoli na základě m/z, ale podle fragmentačních obrazů („patterns ). Ve spojení s chromato či elfo metodou slouží MS jako detektor s vysokým rozlišením. Taková on-line analýza pak dává okamžité výsledky na rozdíl od off-line detekce. Klíčové je propojení přístrojů. LC-MS v biochemii pro peptidy a proteiny, GC-MS pro těkavé LMW látky, CE-MS široký záběr od LMW po proteiny, DNA, viry etc.")
30
Analýza iontů uvolněných ionizací - používané analyzátory
1. Double focusing magnetic sector analyser (analyzátor s dvojím zaostřením) - nejdříve elektrostatická separace podle náboje, následuje separace v magnetickém poli podle hmoty. Výsledkem je tedy separace podle poměru m/z. Elektrické pole: z.e.U = mv2/2 Magnetické pole: B.z.e.v = mv2/r odtud: m/z = (B.r2.e)/2U část: magnetický analyzátor
 - nejdříve elektrostatická separace podle náboje, následuje separace v magnetickém poli podle hmoty. Výsledkem je tedy separace podle poměru m/z. Elektrické pole: z.e.U = mv2/2. Magnetické pole: B.z.e.v = mv2/r. odtud: m/z = (B.r2.e)/2U. část: magnetický analyzátor.")
31
2. Quadrupole mass analyser (kvadrupólový analyzátor) - dva páry paralelních tyčí po stranách iontového paprsku. Změnou stejnosměrného napětí (DC) přiváděného současně s radiofrekvenčním (RF) napětím je možné vybrat látky s určitou hodnotou m/z. zbytek iontů není detekován

32
2a. Quadrupole ion trap IT má velikost tenisového míčku, tři hyperbolické elektrody - prstenec a dvě koncové čepičky. Ionty z ionizátoru jsou zaostřeny do iontové pasti plněné heliem. Díky napěťovým pulsům je IT otevřena (-V) nebo zavřena (+V). Diskontinuální průchod iontů. oscilační napětí
 nebo zavřena (+V). Diskontinuální průchod iontů. oscilační napětí.")
33
3. linear TOF analyser („time of flight“, přeletový) - ionty jsou urychlovány v lineární trubici, bez magnetického pole. Hodnota poměru m/z může být určena z času, který je nutný pro překonání trubice v délce L. TOF analýza se zvláště využívá pro ionizační techniky, které poskytují ionty diskont. (MALDI). 4. TOF reflectron (iontový odražeč, zrcadlo) - kombinace TOF s elektrostatickým analyzátorem, reflektronem. Čas letu je prodloužen, klesá distribuce kinetické energie, tím i časová distribuce (snižuje se šířka píku). Tím se zvyšuje rozlišení. Omezení, vhodné pro m/z menší než 10,000. 5. Fourier transform ion cyclotron resonance analyzer (FT ICR) - částice rotují v magnetickém poli, působením rezonanční radiové frekvence jsou excitovány a tím pádem vzniká proud. Ten může být matematicky transformován, získají se frekvence komponent, které jsou ve vztahu k jejich m/z.
 - kombinace TOF s elektrostatickým analyzátorem, reflektronem. Čas letu je prodloužen, klesá distribuce kinetické energie, tím i časová distribuce (snižuje se šířka píku). Tím se zvyšuje rozlišení. Omezení, vhodné pro m/z menší než 10, Fourier transform ion cyclotron resonance analyzer (FT ICR) - částice rotují v magnetickém poli, působením rezonanční radiové frekvence jsou excitovány a tím pádem vzniká proud. Ten může být matematicky transformován, získají se frekvence komponent, které jsou ve vztahu k jejich m/z.")
34
akcelerace v el. poli: z.e.U = mv2/2 doba letu: t = L/v = L . sqrt (m/2.z.U) m/z = (2. U .t2)/L2
 m/z = (2. U .t2)/L2")
35
Triple quadrupole mass spectrometer (QqQ)
MS/MS v jednom přístroji Q1, filtrace iontů, typický kvadrupólový analyzátor; výběr iontů pro vstup do druhého kvadrupólu q2, neslouží k separaci iontů (fixní RF napětí) - „tunel“, použití jako kolizní cela pro fragmentaci iontů (kolizní plyn, el. pole) Q3, druhá filtrace iontů vycházejících z q2 (u Q TRAP MS instrumentů má funkci lineární iontové pasti) nahrazením Q3 TOF analyzátorem vzniká Qq TOF (QSTAR)
 - „tunel , použití jako kolizní cela pro fragmentaci iontů (kolizní plyn, el. pole) Q3, druhá filtrace iontů vycházejících z q2 (u Q TRAP MS instrumentů má funkci lineární iontové pasti) nahrazením Q3 TOF analyzátorem vzniká Qq TOF (QSTAR)")
36
ESI Qq TOF instrument Q0 Q1 q2

37
MALDI TOF/TOF Nejnovější MS/MS přístroj s MALDI ionizací, konkurent pro ESI Qq TOF a ESI Q TRAP. Kolizní kvadrupól nahrazen TOFem s kolizí (target gas), analýza kolmým TOF analyzátorem s reflektronem 4700 Proteomics Analyzer, Applied Biosystems
, analýza kolmým TOF analyzátorem s reflektronem Proteomics Analyzer, Applied Biosystems.")
38
Electron multiplier - detektor
Tvar rohu, aplikuje se vysoké napětí, opačně vůči iontům, které mají být detekovány (nejč. -V). Kolizemi s povrchem EM se tvoří sekundární elektrony, až 108 po zásahu jedním elektronem. Analogicky funguje fotonásobič, zde jde však o emisi sekundár. fotonů.
. Kolizemi s povrchem EM se tvoří sekundární elektrony, až 108 po zásahu jedním elektronem. Analogicky funguje fotonásobič, zde jde však o emisi sekundár. fotonů.")
39
Analýza primární struktury proteinů (sekvencování) technikou MALDI MS
1. Peptide mass fingerprinting Vzorek proteinu je podroben enzymové nebo chemické degradaci, která je cílena na určitá místa v molekule („site-specific“). Vznikne tak směs peptidů, která je pak analyzována MS. Vhodnou technikou je MALDI TOF MS, neboť se jedná o poměrně složitou směs. Užívá se však i ESI TOF MS. Dnes již klasickou metodou je dřívější zdlouhavý postup, tedy separace peptidů HPLC a následná sekvenční analýza Edmanovou degradací. Sekvenční analýza proteinů s použitím MALDI TOF MS není založena na přesné identifikaci pořadí aminokyselin, ale na získání souboru dat pro vyhledávání v databázích.
. Vznikne tak směs peptidů, která je pak analyzována MS. Vhodnou technikou je MALDI TOF MS, neboť se jedná o poměrně složitou směs. Užívá se však i ESI TOF MS. Dnes již klasickou metodou je dřívější zdlouhavý postup, tedy separace peptidů HPLC a následná sekvenční analýza Edmanovou degradací. Sekvenční analýza proteinů s použitím MALDI TOF MS není založena na přesné identifikaci pořadí aminokyselin, ale na získání souboru dat pro vyhledávání v databázích.")
40
Příprava vzorku vyříznout proužek rozřezat omýt (redukce) (alkylace)
štěpení proteasou Příprava vzorku proč redukce a alkylace? rozvolnění S-S vazeb denaturace > lepší proteolýza ochrana oxidovaných reziduí Cys trypsin agresivní proteasa vysoká primární specificita, nízká sekundární prům. m/z fragmentu ~ 1.5 kDa štěpí: arginyl-X, lysyl-X; ale ne X=prolyl DTT, dithiothreitol KONTAMINACE! – keratiny (kůže, vlasy, srst, vlna) – detergenty (SDS, Triton atp.) IAA, jódoctová kys.
 – detergenty (SDS, Triton atp.) IAA, jódoctová kys.")
41
Princip ESI TOF MS a MALDI TOF MS

42
Hledáním v databázi je pak proteinový vzorek identifikován, nebo se zjistí, že jde o protein, který dosud v databázi není. Předpodkladem pro vyhledávání jsou naměřené přesné m/z hodnoty peptidových fragmentů, použítí specifické proteasy a přesná m/z intaktního proteinu. Pro štěpení se používají endoproteasy, tedy proteolytické enzymy, které štěpí uvnitř proteinové molekuly. Důraz se klade na specifitu použité proteasy, používají se proto vysoce specifické enzymy, jako je trypsin z hovězího či vepřového pankreatu (EC ) nebo Lys C proteasa z Bacillus licheniformis (EC ). Trypsin štěpí za bazickými zbytky Arg a Lys, proteasa Lys C za zbytkem Lysinu. Podobně Glu C proteasa ze Staphylococcus aureus V8 (EC ) štěpí za Glu. Specificky lze štěpit chemicky použitím CNBr.
 nebo Lys C proteasa z Bacillus licheniformis (EC ). Trypsin štěpí za bazickými zbytky Arg a Lys, proteasa Lys C za zbytkem Lysinu. Podobně Glu C proteasa ze Staphylococcus aureus V8 (EC ) štěpí za Glu. Specificky lze štěpit chemicky použitím CNBr.")
43
Bioinformatika pro MS organismy se sekvenovaným genomem
PMF, statistická shoda predikovaných m/z tryptických peptidů (in silico digest) s experimentálními PC programy MASCOT (firma Matrixscience), SEQUEST, ProteinProspector organismy s nesekvenovaným genomem hledání na základě evoluční homologie proteinových sekvencí MS BLAST: MS driven Best Local Alignment Search Tool MultiTag (S. Sunyaev et al., Anal. Chem. 2003, in press)
 s experimentálními. PC programy MASCOT (firma Matrixscience), SEQUEST, ProteinProspector. organismy s nesekvenovaným genomem. hledání na základě evoluční homologie proteinových sekvencí. MS BLAST: MS driven Best Local Alignment Search Tool. MultiTag (S. Sunyaev et al., Anal. Chem. 2003, in press)")
44
Některé proteinové databáze na internetu

45
Peptide Mass Fingerprinting - PMF, MALDI TOF MS
masses (m/z) ELSDIAR QLLLTADDR PHSHPALTPEQK PHSHPALTPEQKK GILAADESTGSIAKR LQSIGTENTEWENRR IGENHTPSALAIMENANVLAR YTPSGQAGAAASESLFISNHAY
 ELSDIAR QLLLTADDR PHSHPALTPEQK PHSHPALTPEQKK GILAADESTGSIAKR LQSIGTENTEWENRR IGENHTPSALAIMENANVLAR YTPSGQAGAAASESLFISNHAY.")
46
Identifikace Glukosa-6-fosfát dehydrogenasy

47
Identifikace hovězího sérového albuminu

48
Identifikace fosforylasy glykogenu

49
Někdy může navazovat použití specifických exoproteas a zjišťovat tak koncové aminokyseliny.

50
In-silico sequencing Počítačová predikce štěpných peptidů určitého proteinu. Výhodné použití pro usnadnění identifikace v analýze PMF v případě, že je vzorek znečištěn jinými proteiny. Rozumné použití pro max. 3 proteiny v jednom vzorku. Zadáním parametrů v PC programu např. Proteogest (použitý proces štěpení - CNBr, trypsin; výskyt posttranslačních modifikací etc.) se získá příslušný datový soubor
 se získá příslušný datový soubor.")
51
Př. distribuce urč. aminokyselin v lidském proteomu

52
2. Post source decay (PSD), doplněk k PMF
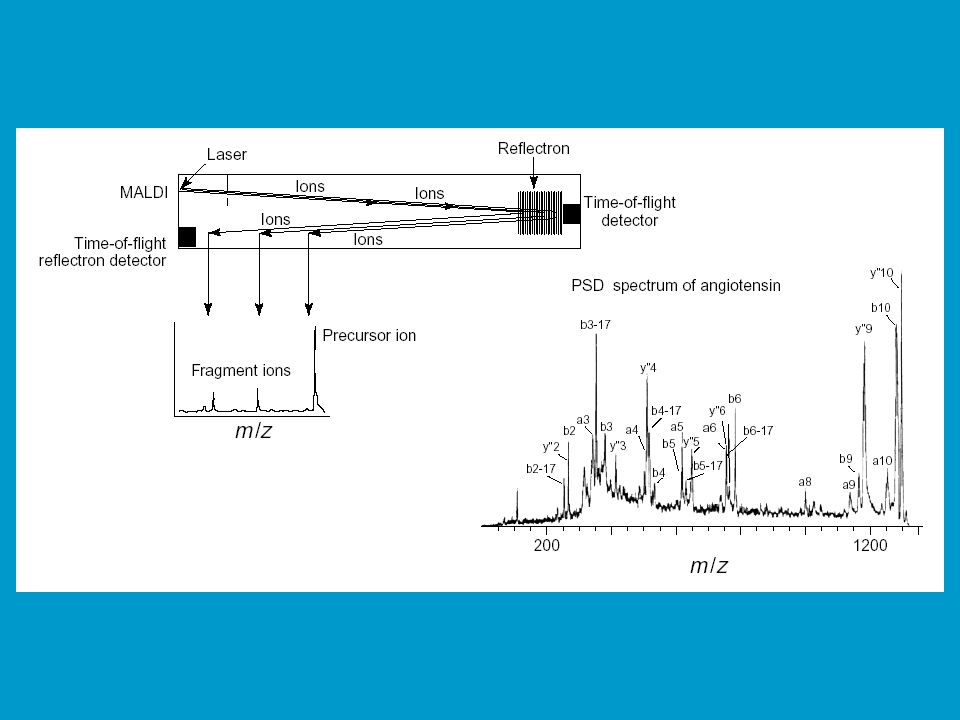
Fragmentace iontů mezi iontovým zdrojem a detektorem (rozpad v letové trubici), tedy v místech, kde nepůsobí žádné silové pole. K fragmentaci dochází ve fázi desorpce a akcelerace, díky kolizím mezi ionty analytu a dalšími molekulami (tzv. residual gas - např. neutrální molekuly matrix). Ionizuje se při zvýšené energie laseru ve srovnání s normální MALDI. Na počátku mají tyto fragmenty stejnou rychlost jako mateřský ion. V lineárním TOF analyzátoru proto dorazí k detektoru ve stejném čase, neobjeví se proto v hmotnostním spektru. Při použití TOF reflektronového analyzátoru tyto fragmenty vnikají do nitra reflektronu, do různé hloubky (podle velikosti kinetické energie, tím pádem pak doletí do reflektronového detektoru v různých časech. Peptide fragment sequencing - jde o velmi populární metodu pro sekvencování peptidů.
, tedy v místech, kde nepůsobí žádné silové pole. K fragmentaci dochází ve fázi desorpce a akcelerace, díky kolizím mezi ionty analytu a dalšími molekulami (tzv. residual gas - např. neutrální molekuly matrix). Ionizuje se při zvýšené energie laseru ve srovnání s normální MALDI. Na počátku mají tyto fragmenty stejnou rychlost jako mateřský ion. V lineárním TOF analyzátoru proto dorazí k detektoru ve stejném čase, neobjeví se proto v hmotnostním spektru. Při použití TOF reflektronového analyzátoru tyto fragmenty vnikají do nitra reflektronu, do různé hloubky (podle velikosti kinetické energie, tím pádem pak doletí do reflektronového detektoru v různých časech. Peptide fragment sequencing - jde o velmi populární metodu pro sekvencování peptidů.")
53
Schéma instrumentu pro MALDI PSD TOF MS

55
de novo sequencing MALDI PSD TOF MS (LC)-ESI-QqTOF MS
-ESI-QqTOF MS")
56
3. Kombinace MALDI s MSn a analýzou FT
Disociace vybraného mateřského iontu je dosaženo díky koliznímu plynu nebo použitím pulsních laserů (fotodisociace). Výhodou je vysoké rozlišení, přesnost a citlivost. Fragmentace ovšem poskytuje pouze omezené množství iontů.
. Výhodou je vysoké rozlišení, přesnost a citlivost. Fragmentace ovšem poskytuje pouze omezené množství iontů.")
57
Charakterizace ko- a posttranslačních modifikací
v proteinech s použitím MS fosforylace, sulfatace, glykosylace, N-koncové modifikace Význam: důležité pro regulaci buněčné distribuce a modulaci funkcí proteinů. Př. fosforylace - přenos signálu v buňce, glykosylace - vliv na funkční, strukturní a imunologické vlastnosti proteinů. Výhoda MALDI MS analýzy v tomto případě spočívá ve velké přesnosti určení molekulové hmotnosti, citlivosti a rozlišení. Typický protokol pro analýzu proteinové modifikace začíná štěpením vzorku. Pak se analyzuje rozdíl v molekulové hmotnosti určitého peptidu spočítané na základě sekvence a odečtené z MS spektra. Pro složitost některých směsí je výhodnější použít MALDI PSD MS/MS nebo LC MS.

59
MALDI MS pro studium vyšší struktury proteinů
Studium prostorové struktury proteinů (zvl. terciární a kvarterní) má význam z důvodu těsného vztahu struktura - funkce. Poznání struktury tedy může poskytnout informace o funkci proteinu. Pro studium struktury proteinů se běžně používá řada spektro-skopických metod (Krystalografie - difrakce paprsků X, NMR spektroskopie a další). Dávají poměrně jasné výsledky, ale jejich nevýhodou může být zejména časová náročnost a nároky na vyšší množství materiálu. Poskytují rovněž značná množství dat, jejichž vyhodnocování je opět náročné. Technikou MS je možné studovat například přístupnost určitých povrchových regionů proteinů s použiím metody zvané protein mass fingerprinting (PMF).
 má význam z důvodu těsného vztahu struktura - funkce. Poznání struktury tedy může poskytnout informace o funkci proteinu. Pro studium struktury proteinů se běžně používá řada spektro-skopických metod (Krystalografie - difrakce paprsků X, NMR spektroskopie a další). Dávají poměrně jasné výsledky, ale jejich nevýhodou může být zejména časová náročnost a nároky na vyšší množství materiálu. Poskytují rovněž značná množství dat, jejichž vyhodnocování je opět náročné. Technikou MS je možné studovat například přístupnost určitých povrchových regionů proteinů s použiím metody zvané protein mass fingerprinting (PMF).")
60
Peptide mass fingerprinting v analýze 3D struktury
Proteolyt. štěpení proteinu může poskytnout rozdílné peptidové mapy v závislosti na momentální prostorové struktuře (konformaci) a tím obnažení (nebo naopak skrytí) míst, kde použitá proteasa štěpí. Příkladem může být protein kalmodulin (CaM), což je alosterický regulační protein, který mění konformace v závislosti na přítomnosti či nepřítomnosti vápníku, který je alosterickým efektorem. Po vazbě vápníku a následné proteolýze tedy dostaneme peptidovou mapu, která se liší od té bez vápníku. Kalmodulin vypadá jako „činka“, kde dva globulární regiony jsou spojeny dlouhým a-helixem.Vazba vápníku způsobí v tomto případě strukturní změnu, která znesnadní štěpení helikální části.
 a tím obnažení (nebo naopak skrytí) míst, kde použitá proteasa štěpí. Příkladem může být protein kalmodulin (CaM), což je alosterický regulační protein, který mění konformace v závislosti na přítomnosti či nepřítomnosti vápníku, který je alosterickým efektorem. Po vazbě vápníku a následné proteolýze tedy dostaneme peptidovou mapu, která se liší od té bez vápníku. Kalmodulin vypadá jako „činka , kde dva globulární regiony jsou spojeny dlouhým a-helixem.Vazba vápníku způsobí v tomto případě strukturní změnu, která znesnadní štěpení helikální části.")
61
PMF - konformace kalmodulinu

62
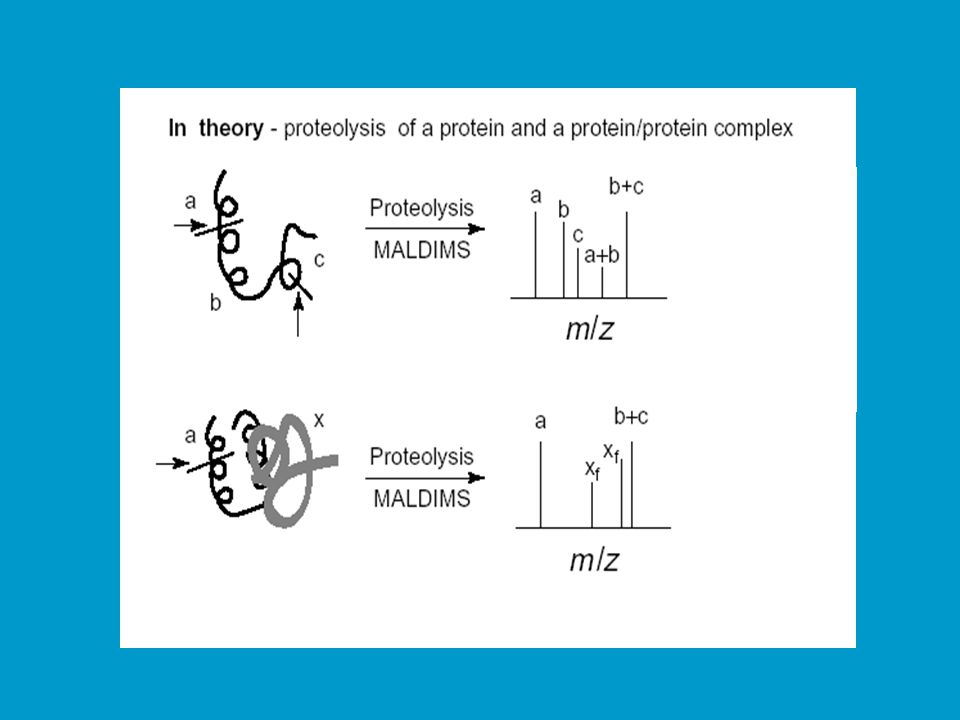
Studium protein/proteinové interakce (kvarterní struktura)
Cílem je studovat regiony, které se podílejí na interakci podjednotek v kvarterní struktuře či interakce více proteinů. Rozdíly v peptidových mapách mohou být vyvolány přímými či nepřímými změnami expozice štěpných míst. Přímá změna: vazbou jiné podjednotky či proteinu dojde ke sterickému zablokování určitých štěpných míst. Nepřímá změna: vazbou jiné podjednotky či proteinu dojde ke konformační změně, která se projeví jinde zakrytím určitého štěpného místa. Jinou možností výzkumu protein-proteinových interakcí (struktur vyššího řádu) je studium chemické reaktivity aminokyselin (acylace, sukcinylace) nebo protein cross-linking (křížová vazba). V druhém případě jde o zesíťování proteinu bifunkčním činidlem před štěpením. Vzniklé fragmenty jsou indikátorem 3D struktury.
 je studium chemické reaktivity aminokyselin (acylace, sukcinylace) nebo protein cross-linking (křížová vazba). V druhém případě jde o zesíťování proteinu bifunkčním činidlem před štěpením. Vzniklé fragmenty jsou indikátorem 3D struktury.")
64
Aplikace LDI MS 1. Diagnostika
MALDI MS nachází uplatnění v tomto oboru díky schopnosti analyzovat složité směsi látek. Biologické tekutiny jako sérum, mozkomíšní mok nebo moč však často obsahují nízké koncentrace studovaných analytů. Podobně i nadbytek některých proteinů (např. hemoglobin v séru) často znesnaňuje vyšetřování („screening“). Je proto nutné primárně analyt izolovat (zakoncentrovat) a teprve potom podrobit analýze MALDI MS. Mass spectrometric immunoassay (MSIA) spoléhá na afinitní záchyt analytu immobilizovanou protilátkou. Tímto způsobem je možné detekovat femtomolární množství (nanomolární koncentrace).
 často znesnaňuje vyšetřování („screening ). Je proto nutné primárně analyt izolovat (zakoncentrovat) a teprve potom podrobit analýze MALDI MS. Mass spectrometric immunoassay (MSIA) spoléhá na afinitní záchyt analytu immobilizovanou protilátkou. Tímto způsobem je možné detekovat femtomolární množství (nanomolární koncentrace).")
65
MSIA Analyt je prokázán podle specifické hodnoty m/z
kyselá hydrolýza vazby Analyt je prokázán podle specifické hodnoty m/z

66
hmotová spektrometrie
SELDI – surface enhanced laser desorption/ionisation hmotová spektrometrie array chip výhody jednoduchost rychlost selektivita nevýhody nízká citlivost nízká přesnost urč. m/z nízké rozlišení retentát

68
2. Automatizované kvantitativní analýzy s MALDI MS
Počítačem řízené sériové analýzy (multisampler). Iontový signál je monitorován v závislosti na změnách v umístění laseru (spirálovitě od středu spotu) a jeho intenzitě. Tyto alterace probíhají dokud signál nedosáhne potřebné intenzity (předvolené) nad prahovou hodnotu (threshold). Pak přesun na následující spot. Pro kvantitativní analýzy nutná kalibrace. Př. detekce toxinů v tělních tekutinách.
. Iontový signál je monitorován v závislosti na změnách v umístění laseru (spirálovitě od středu spotu) a jeho intenzitě. Tyto alterace probíhají dokud signál nedosáhne potřebné intenzity (předvolené) nad prahovou hodnotu (threshold). Pak přesun na následující spot. Pro kvantitativní analýzy nutná kalibrace. Př. detekce toxinů v tělních tekutinách.")
69
3. One-step analýza imobilizovaných peptidů a oligosacharidů
Imobilizované biomolekuly jsou odštěpeny z matrice (polystyren, umělé pryskyřice) po zásahu kvantem energie laseru. Podmínkou je připojení k pevné fázi prostřednictvím fotolabilního linkeru. Použití pro studium reakcí na pevné fázi a pro evaluaci purifikačních metod využívajících pevné fáze.
 po zásahu kvantem energie laseru. Podmínkou je připojení k pevné fázi prostřednictvím fotolabilního linkeru. Použití pro studium reakcí na pevné fázi a pro evaluaci purifikačních metod využívajících pevné fáze.")
Podobné prezentace














>")
 REZONANCE>")





