Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Molekulární diagnostika vrozených nemocí
Lenka Fajkusová Centrum molekulární biologie a genové terapie Fakultní nemocnice Brno Černopolní 9 Brno

2
Molekulární problematika dědičných
neuromuskulárních nemocí metabolických nemocí kožních nemocí nemocí pojivových a kostních tkání mentálních retardací

3
Neuromuskulární nemoci
Disease Gene Localization Protein Duchenne/Becker muscular dystrophy DMD Xp21 Dystrophin Spinal muscular atrophy SMN1 5q12 Survival motor neuron protein 1 Myotonic dystrophy, type 1 DMPK 19q13 Dystrophia myotonia protein kinase Myotonic dystrophy, type 2 ZNF9 3q13 Zinc finger protein 9 Facioscapulohumeral muscular dystrophy, type 1 Deletion of D4Z4 repeats 4q35 Non-dystrophic myotonia CLCN1, SCN4A 7q35, 17q23 Muscle chloride channel 1, Sodium channel 4A Limb girdle muscular dystrophy, type 2A, 2D, 2I, 2L CAPN3, SGCA, FKRP, ANO5 15q15, 17q12, 19q13, 11p14 Calpain 3, Sarcoglycan alpha; Fukutin-related protein, Anoctamin 5 Sequence capture and targeted resequencig 42 genes

4
Metabolické nemoci Disease Gene Localization Protein
Familial hypercholesterolemia LDLR 19p13 Low density lipoprotein receptor Familial hypercholesterolemia, type B APOB 2p24 Apolipoprotein B100 Phenylketonuria PAH 12q24 Phenylalanine hydroxylase Congenital adrenal hyperplasia CYP21A2 6p21 21-hydroxylase Alpha-1-antitrypsin deficiency PI 14q32 Alpha-1-antitrypsin Smith-Lemli-Opitz syndrome DHCR7 11q12 Sterol delta-7-reductase Wilson disease ATP7B 13q14 ATPase, Cu(2+)-transporting beta polypeptide Galactosemia GALT 9p13 Galactose-1-phosphate uridylyltransferase Glycogen storage disease, type 1A G6PC 17q21 Glucose-6-phosphatase Glycogen storage disease, type 1B G6PT 11q23 Glucose-6-phosphate transporter protein Glycogen storage disease, type 2 GAA 17q25 Acid alpha-1,4-glucosidase Glycogen storage disease, type 3 AGL 1p21 Glycogen debrancher enzyme
-transporting beta polypeptide. Galactosemia. GALT. 9p13. Galactose-1-phosphate uridylyltransferase. Glycogen storage disease, type 1A. G6PC. 17q21. Glucose-6-phosphatase. Glycogen storage disease, type 1B. G6PT. 11q23. Glucose-6-phosphate transporter protein. Glycogen storage disease, type 2. GAA. 17q25. Acid alpha-1,4-glucosidase. Glycogen storage disease, type 3. AGL. 1p21. Glycogen debrancher enzyme.")
5
Kožní nemoci Disease Gene Localization Protein, function
Epidermolysis bullosa simplex KRT5, KRT14, PLEC 12q13, 17q12, 8q24 Keratin 5, Keratin 14, Plectin Epidermolysis bullosa dystrophica COL7A1 3p21 Type VII collagen, alpha-1 chain Incontinentia pigmenti NEMO Xq28 NF-kappa-B essential modulator Ichthyoses FLG, STS, TGM1, ALOX12B, ALOXE3, NIPAL4, CYP4F22 1q21, Xp22, 14q12, 17p13, 17p13, 5q33, 19p13 Filaggrin, Steroid sulphatase, Transglutaminase 1, 12R-lipoxygenase, Lipoxygenase 3, Nipal-like domain-containing 4 (ichthyin), Cytochrome P450
, Cytochrome P450.")
6
Onemocnění …. asociovaný gen …. kódovaný protein …. funkce proteinu …
Onemocnění ….. asociovaný gen ….. kódovaný protein ….. funkce proteinu ….. vliv mutace/mutací na funkci proteinu ….. molekulární podstata onemocnění ….. klinické projevy onemocnění ….. používané metody DNA diagnostiky

7
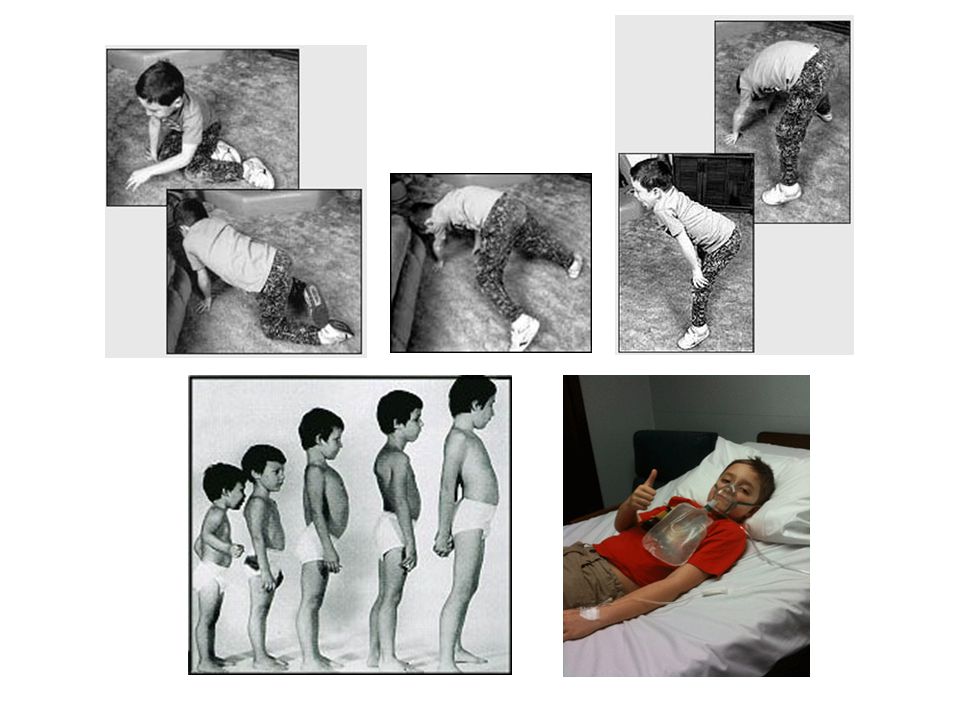
Svalové dystrofie - progresivní slabost a atrofie kosterních svalů
Diagnóza svalové dystrofie je založena na klinických nálezech, histopatologické analýze svalové tkáně, genetické analýze. Ztráta svalové hmoty nemusí být zřetelná - svaly jsou nahrazovány tukovou a pojivovou tkání - pseudohypertrofie. U různých typů svalových dystrofií jsou postiženy specifické svalové skupiny, zvláště v počátku onemocnění.

8
Guillaume-Benjamin Duchenne de Boulogne
1868: De la Paralysie Musculaire Pseudo-hypertrophique...

10
Duchennova svalová dystrofie (DMD)
Incidence: 1/3500 chlapců; mutace v genu pro dystrofin (Xp21); dystrofin - protein membránového skeletu svalových buněk. Imunohistochemická analýza dystrofinu v tkáňových řezech Western blotting
; dystrofin - protein membránového skeletu svalových buněk. Imunohistochemická analýza dystrofinu. v tkáňových řezech. Western blotting.")
11
Izolace DNA (leukocyty)
Molekulárně genetická diagnostika: analýza DNA Analýza 79 exonů metodou MLPA - Multiple Ligation dependent Probe Amplification Izolace DNA (leukocyty) Delece/duplikace není detekována Analýza jednotlivých exonů na přítomnost bodových mutací (PCR-sekvenování) Delece/duplikace je detekována Určení přenašečství delece/duplikace v rodině
 Delece/duplikace není detekována. Analýza jednotlivých exonů na přítomnost bodových mutací (PCR-sekvenování) Delece/duplikace je detekována. Určení přenašečství delece/duplikace v rodině.")
12
Detekce rozsáhlých delecí/duplikací – Multiplex Ligation-dependent Probe Amplification
Denatured genomic DNA is hybridised with a mixture of probes. Each MLPA probe consists of two oligonucleotides. Oligonucleotides hybridise to adjacent target sequences and are ligated by a thermostable ligase. All probe ligation products are amplified simultaneously by PCR using a single primer pair. The amplification product of each probe has a unique length. Amplification products are separated by capillary electrophoresis. Relative amounts of probe amplification products reflect the relative copy number of target sequences.

13
Reverzní transkripce, amplifikace (10 fragmentů)
Molekulárně genetická diagnostika: analýza mRNA Izolace RNA (svalové buňky) Reverzní transkripce, amplifikace (10 fragmentů) Delece/duplikace není detekována Elektroforéza Protein truncation test - specifická detekce nonsense a frame-shift mutací Delece/duplikace je detekována M K P1 P2 P3 P4
 Reverzní transkripce, amplifikace (10 fragmentů) Delece/duplikace není detekována. Elektroforéza. Protein truncation test - specifická detekce nonsense a frame-shift mutací. Delece/duplikace je detekována. M K P1 P2 P3 P4.")
14
Protein truncation test
mRNA cDNA Reverzní transkripce 1. PCR 2. PCR T7 ATG In vitro transkripce/translace TAG PCR produkt RNA Protein K P

15
Pacient s DMD: RT-PCR DNA analýza (MLPA, sekvenční analýza exonů a přilehlých intronových oblastí: mutace nezjištěna mRNA analýza (RT, PCR; sekvenční analýza): exon 65 – 53 nt. intronu 65 – exon 66 DNA analýza: c T>G; změna sestřihu mRNA, inzerce pseudoexonu Analýza DNA T>G
: exon 65 – 53 nt. intronu 65 – exon 66. DNA analýza: c T>G; změna sestřihu mRNA, inzerce pseudoexonu. Analýza DNA. T>G.")
16
Imunodetekce dystrofinu pomocí protilátky DYS2 ve svalové tkáni
Duchennova svalová dystrofie - nonsense mutace, delece a duplikace měnící čtecí rámec translace Beckerova svalová dystrofie - delece a duplikace zachovávající čtecí rámec translace Imunodetekce dystrofinu pomocí protilátky DYS2 ve svalové tkáni Normální vzorek Pacient s BMD Western blot Pacient s DMD

17
Pacient s BMD nesoucí nonsense mutaci Glu1110X
P K M C(3536)>A; exon 25; Glu1110X PTT: 1. Kontrola, 2. BMD pacient Detekce transkriptu s alternativním sestřihem exonu 25 ( del) transkript s in-frame delecí RT-PCR
>A; exon 25; Glu1110X. PTT: 1. Kontrola, 2. BMD pacient. Detekce transkriptu s alternativním sestřihem exonu 25 ( del) transkript s in-frame delecí. RT-PCR.")
18
a | The essential splicing signals that define the exon boundaries - GU and AG dinucleotides, the branch-point adenosine, polypyrimidine tract of variable length. Components of the basal splicing machinery bind to the consensus sequences and promote assembly of the splicing complex. The U1 snRNP binds to the 5'-splice site, and the U2 snRNP binds the branch site through RNA–RNA interactions. Additional enhancer and silencer elements in the exons and introns (ESE, ESS, ISE, ISS) allow the correct splice sites to be distinguished from the many cryptic splice sites that have identical signal sequences. Trans-acting splicing factors can interact with enhancers and silencers and can accordingly be subdivided into two main groups: members of the SR family of proteins and of the hnRNPs. In general, SR protein binding at ESE facilitates exon recognition whereas hnRNPs are inhibitory. Protein–protein interactions in the spliceosome that modulate the recognition of the splice sites are the probable cause of splicing inhibition or activation. b | Genomic variants (GVs) can affect different splicing regulatory elements, leading to aberrant splicing. Nature Reviews Genetics 5,

19
a | The essential splicing signals that define the exon boundaries are relatively short and poorly conserved sequences. Only the GU and the AG dinucleotides that directly flank the exon (at the 3' and 5' ends, respectively) and the branch-point adenosine (all in red) are always conserved. In most cases, there is also a polypyrimidine tract of variable length (the consensus symbol 'y' represents a pyrimidine base — cytosine or thymine) upstream of the 3'-splice site. The branch point is typically located 18–40 nucleotides upstream from the polypyrimidine tract. Components of the basal splicing machinery bind to the consensus sequences and promote assembly of the splicing complex. This multiprotein complex, known as a spliceosome, performs the correct identification of the splicing signals and catalysis of the cut-and-paste reactions (Fig. 1). Five small nuclear ribonucleoproteins (snRNPs) and more than 100 proteins make up the spliceosome. The U1 snRNP binds to the 5'-splice site, and the U2 snRNP binds the branch site through RNA–RNA interactions. Additional enhancer and silencer elements in the exons (EXON SPLICING ENHANCER (ESE); EXON SPLICING SILENCER (ESS)) and/or introns (INTRON SPLICING ENHANCER (ISE); INTRON SPLICING SILENCER (ISS)) allow the correct splice sites to be distinguished from the many cryptic splice sites that have identical signal sequences. Trans-acting splicing factors can interact with enhancers and silencers and can accordingly be subdivided into two main groups: members of the serine arginine (SR) family of proteins and of the HETEROGENEOUS NUCLEAR RIBONUCLEOPROTEIN PARTICLES (hnRNPs). In general, SR protein binding at ESE facilitates exon recognition whereas hnRNPs are inhibitory. Protein–protein interactions in the spliceosome that modulate the recognition of the splice sites are the probable cause of splicing inhibition or activation. b | Genomic variants (GVs) can affect different splicing regulatory elements, leading to aberrant splicing. Exonic GVs (eGVs) can either change the amino acid, result in synonymous GVs in exons (sGVs) or introduce a nonsense codon. Intronic GVs might be located within approximately 50 bp from the splice sites (that is, 3'-splice site GVs (ssGVs) and 5' ssGVs) or deep in the introns (intronic GVs (iGVs)).
 and the branch-point adenosine (all in red) are always conserved. In most cases, there is also a polypyrimidine tract of variable length (the consensus symbol y represents a pyrimidine base — cytosine or thymine) upstream of the 3 -splice site. The branch point is typically located 18–40 nucleotides upstream from the polypyrimidine tract. Components of the basal splicing machinery bind to the consensus sequences and promote assembly of the splicing complex. This multiprotein complex, known as a spliceosome, performs the correct identification of the splicing signals and catalysis of the cut-and-paste reactions (Fig. 1). Five small nuclear ribonucleoproteins (snRNPs) and more than 100 proteins make up the spliceosome. The U1 snRNP binds to the 5 -splice site, and the U2 snRNP binds the branch site through RNA–RNA interactions. Additional enhancer and silencer elements in the exons (EXON SPLICING ENHANCER (ESE); EXON SPLICING SILENCER (ESS)) and/or introns (INTRON SPLICING ENHANCER (ISE); INTRON SPLICING SILENCER (ISS)) allow the correct splice sites to be distinguished from the many cryptic splice sites that have identical signal sequences. Trans-acting splicing factors can interact with enhancers and silencers and can accordingly be subdivided into two main groups: members of the serine arginine (SR) family of proteins and of the HETEROGENEOUS NUCLEAR RIBONUCLEOPROTEIN PARTICLES (hnRNPs). In general, SR protein binding at ESE facilitates exon recognition whereas hnRNPs are inhibitory. Protein–protein interactions in the spliceosome that modulate the recognition of the splice sites are the probable cause of splicing inhibition or activation. b | Genomic variants (GVs) can affect different splicing regulatory elements, leading to aberrant splicing. Exonic GVs (eGVs) can either change the amino acid, result in synonymous GVs in exons (sGVs) or introduce a nonsense codon. Intronic GVs might be located within approximately 50 bp from the splice sites (that is, 3 -splice site GVs (ssGVs) and 5 ssGVs) or deep in the introns (intronic GVs (iGVs))..")
20
Phenotype Localisation (exon) Mutation detected at cDNA level DNA level Mutation at protein level Immunohistochemical labelling using antibodies DYS1,2,3 BMD 3 c.[163G>T, 94_649del] Not performed p.[E55X, F32_D217del] The patient has the mutation c.163G>T (p.E55X). Besides transcript with this mutation, transcript with the frame-shift deletion of exons 3–7 (c.94_649del) was detected. Winnard et al. [Am J Hum Genet 1995] described a BMD patient with the deletion of exons 3–7 (antibodies directed against the amino-terminus and the 5´ end of exon 8 did not detect dystrophin in muscles of this patient, but dystrophin was detected with an antibody directed against the 3´ end of exon 8. The authors supposed that dystrophin production is initiated at a new start codon in exon 8, and a similar mechanism could have acted in the case of our patient.
. Besides transcript with this mutation, transcript with the frame-shift deletion of exons 3–7 (c.94_649del) was detected. Winnard et al. [Am J Hum Genet 1995] described a BMD patient with the deletion of exons 3–7 (antibodies directed against the amino-terminus and the 5´ end of exon 8 did not detect dystrophin in muscles of this patient, but dystrophin was detected with an antibody directed against the 3´ end of exon 8. The authors supposed that dystrophin production is. initiated at a new start codon in exon 8, and a similar mechanism could have acted in the case of our patient.")
21
Phenotype Localisation of mutation (exon) Mutation detected at cDNA level DNA level Mutation at protein level Immunohistochemical labelling using antibodies DYS1,2,3 BMD Intron 52 c.[=, 7543_7660del] c delA p.[=, A2515LfsX22] Decreased intensity The patient has the mutation c delA which changes splicing of exon 52 and both transcripts (transcript with frame-shift deletion c.7543_7660del and normal transcript) are generated. Phenotype Localisation (exon) Mutation detected at cDNA level DNA level Mutation at protein level Immunohistochemical labelling using antibodies DYS1,2,3 BMD Intron 33 c.[=, 4674_4675ins10] c A>G p.[=, V1559FfsX20] DYS1 negative, Dys2 and DYS3 decreased intensity The patient has the mutation c A>G which changes splicing and both transcripts are generated (transcript with insertion of a part of intron 33 (c.4674_4675ins10) and normal transcript). aagagtaaactaaattacatttcattataattcttttcagGTAACAGAAAGAAAGCAACAGTTGGA
 are generated. Phenotype. Localisation (exon) Mutation detected at. cDNA level. DNA level. Mutation at protein. level. Immunohistochemical labelling using. antibodies DYS1,2,3. BMD. Intron 33. c.[=, 4674_4675ins10] c A>G. p.[=, V1559FfsX20] DYS1 negative, Dys2 and DYS3 decreased intensity. The patient has the mutation c A>G which changes splicing and both transcripts are generated (transcript with insertion of a part of intron 33 (c.4674_4675ins10) and normal transcript). aagagtaaactaaattacatttcattataattcttttcagGTAACAGAAAGAAAGCAACAGTTGGA.")
22
Phenotype Localisation of mutation (exon) Mutation detected at cDNA level Mutation detected at DNA level Mutation at protein level Immunohistochemical labelling using antibodies DYS1,2,3 BMD 25 c.[3328G>T, 3277_3432del] c.3328G>T p.[E1110X, L1093_Q1144del] Decreased intensity c.3277_3432del c.3432G>A p.L1093_Q1144del DYS1 and DYS2 normal, DYS3 decreased intensity Intron 25 c.[=, 3277_3432del] c G>A p.[=, Normal c T>C DYS1, DYS2 and DYS3 decreased intensity tcatatctaatatgtggcagtaatttttttcagctggcttaaattgatttattttcttagCTTTTAGTCAGTGATATTCAGACAATTCAGCCCAGTCTAAACAGTGTCAATGAAGGTGGGCAGAAGATAAAGAATGAAGCAGAGCCAGAGTTTGCTTCGAGACTTGAGACAGAACTCAAAGAACTTAACACTCAGTGGGATCACATGTGCCAACAGgtatagacaatctctttcactgtggcttgcctcaacgtacttaactaagatttcctaatg P K M

23
Most heterozygous female carriers of DMD mutations are asymptomatic;
however, between 3% and 8% of these carriers are manifesting carriers who develop symptoms ranging from mild muscle weakness to a rapidly progressive DMD-like muscular dystrophy. A B Immunohistochemical staining of dystrophin, A: mosaic pattern of dystrophin expression, but the majority of fibers do not express dystrophin in a DMD manifesting carrier with 100% skewing of X-chromosome inactivation, and B: control skeletal muscle. Neuromuscular Disorders 20 (2010) 499–504
 499–504.")
24
Limb Girdle Muscular Dystrophy, LGMD
LGMD - a group of disorders with wide genetic and clinical heterogeneity 8 types of LGMD with autosomal dominant inheritance (LGMD1A-?) 23 types of LGMD with autosomal recessive inheritance (LGMD2A-?) Patient with α-sarcoglycan (LGMD2D) manifests involvement of the shoulder girdle muscles, (A) scapular winging, scoliosis, and (B) hypertrophy of the calf.
 23 types of LGMD with autosomal recessive inheritance (LGMD2A- ) Patient with α-sarcoglycan (LGMD2D) manifests involvement of the shoulder girdle muscles, (A) scapular winging, scoliosis, and (B) hypertrophy of the calf.")
25
Limb Girdle Muscular Dystrophy, LGMD
The average overall prevalence for all of the AR forms of LGMD (LGMD2) is estimated to range from 1:15,000 to 1:100,000, and this can vary extensively between populations.
 is estimated to range from. 1:15,000 to 1:100,000, and this can vary extensively between populations.")
26
The most common form of LGMD LGMD2B DYSF, Dysferlin
Muscular dystrophy Gene, protein Relative prevalence LGMD2A CAPN3, Calpain-3 The most common form of LGMD LGMD2B DYSF, Dysferlin More common in southern that northen Europe LGMD2C SGCG, Gamma-sarcoglycan Present worldwide LGMD2D SGCA, Alpha-sarcoglycan Present worldwide, the most frequent sarcoglycanopathy LGMD2E SGCB, Beta-sarcoglycan Indiana Amish LGMD2F SGCD, Delta-sarcoglycan African-Brazilian LGMD2G TCAP, Telethonin Brazil LGMD2H TRIM32, TRIM32 Hutterite population of Canada LGMD2I FKRP, Fukutin-related protein Relative frequent in northern Europe LGMD2J TTN, Titin Finland LGMD2K POMT1, O-manosyl transferase-1 Few reported cases (Turkish and English families) LGMD2L FKTN, Fukutin Few reported cases LGMD2M, LGMD2N POMGn1, POMT2 ANO5, Anoctamin 5
 LGMD2L. FKTN, Fukutin. Few reported cases. LGMD2M, LGMD2N. POMGn1, POMT2. ANO5, Anoctamin 5.")
27
Svalové vlákno (počet ve svalu 10 tisíc – 1 milion, délka cm až dm, průměr 10 – 100 µm).
Myofibrila, svalové vlákénko, základní kontraktilní jednotka svalové buňky. Stavba myofibril způsobuje příčně pruhovaný vzhled svalu (tmavé a světlé pruhy). Ten vzniká uspořádáním dvou druhů bílkovinných vláken - aktinových filament a myosinových filament. Aktinová vlákna jsou zakotvena v Z-linii, rozdělující myofibrily v jednotlivé sarkomery, úseky dlouhé v klidu cca 2,2 – 2,8 µm. Sarkomera tedy leží mezi dvěma Z-liniemi. V blízkosti Z-linie tvoří sarkomeru pouze aktinová filamenta, tato část se nazývá I-proužek. Směrem ke středu sarkomery se nachází A-proužek, kde se překrývají aktinová a myosinová vlákna. O něco světlejší středová část sarkomery obsahující jen myosinová vlákna se označuje jako H-proužek (linie H). Jednotlivé linie a proužky se v myofibrile střídají, čímž vzniká vzhled pruhování. Při stahu svalu (kontrakci svalu) se délka sarkomery zkracuje (aktinová a myosinová filamenta se do sebe vzájemně zasouvají).
. Ten vzniká uspořádáním dvou druhů bílkovinných vláken - aktinových filament a myosinových filament. Aktinová vlákna jsou zakotvena v Z-linii, rozdělující myofibrily v jednotlivé sarkomery, úseky dlouhé v klidu cca 2,2 – 2,8 µm. Sarkomera tedy leží mezi dvěma Z-liniemi. V blízkosti Z-linie tvoří sarkomeru pouze aktinová filamenta, tato část se nazývá I-proužek. Směrem ke středu sarkomery se nachází A-proužek, kde se překrývají aktinová a myosinová vlákna. O něco světlejší středová část sarkomery obsahující jen myosinová vlákna se označuje jako H-proužek (linie H). Jednotlivé linie a proužky se v myofibrile střídají, čímž vzniká vzhled pruhování. Při stahu svalu (kontrakci svalu) se délka sarkomery zkracuje (aktinová a myosinová filamenta se do sebe vzájemně zasouvají).")
28
LGMD2A, Calpain 3, CAPN3 CAPN3: calcium-dependent protease, do not completely demolish substrates but cleave them in a controlled and limited manner. CAPN3 is the first enzyme described in association with muscular dystrophy. Majority of CAPN3 is anchored to the cytoskeleton of sarcomere, bound to the gigantic protein titin.

29
Anchoring to titin has a triple function:
LGMD2A, Calpain 3, CAPN3 Anchoring to titin has a triple function: to keep CAPN3 from autolytically degrading itself, to maintain it in a proteolytically inactive state, to place it in proximity of its substrates.

30
LGMD2A, Calpain 3, CAPN3 CAPN3 main roles - to rid the cell of damaged sarcomeric proteins and in this way contribute to maintenance of effectively functioning muscle. E3 ligase is the final enzyme in the process of adding polyubiquitin chains to protein substrates, targeting them to the proteasome. Compatible with the fact that the E3 ubiquitin ligase TRIM32, when mutated can also give rise to LGMD2H. Neuromuscul Disord. 2008; 18(12): 913–921
: 913–921.")
31
Limb Girdle Muscular Dystrophy, LGMD
LGMD diagnostics is based on clinical findings and results of pathological analysis of muscle tissue Western blotting of calpain-3; 1, 2: normal control, 3 - 6: absence of calpain-3 on 94kDa in LGMD2A patients. (FNB, PAU, Hermanová M.) Immunodetection of alpha-sarcoglycan; A: normal control, B: deficiency of alpha-sarcoglycann in a LGMD2D patient. (FNB, PAU, Hermanová M.) A B
 Immunodetection of alpha-sarcoglycan; A: normal control, B: deficiency of alpha-sarcoglycann in a LGMD2D patient. (FNB, PAU, Hermanová M.) A. B.")
32
DNA diagnostics LGMD2 in CMBGT:
Limb Girdle Muscular Dystrophy, LGMD DNA diagnostics LGMD2 in CMBGT: 218 patients, in 86 identified mutations (39,5%) LGMD2A – CAPN3 – sequencing analysis (24 exons); 71 patients LGMD2D – SGCA – sequencing analysis (10 exons); 3 patients LGMD2I – FKRP – detection of the most common mutation p.Leu276Ile; 9 patients LGMD2L – ANO5 – detection of the most common mutation c.191dupA; 3 patients Sequence capture – targeted resequencing
 LGMD2A – CAPN3 – sequencing analysis (24 exons); 71 patients. LGMD2D – SGCA – sequencing analysis (10 exons); 3 patients. LGMD2I – FKRP – detection of the most common mutation p.Leu276Ile; 9 patients. LGMD2L – ANO5 – detection of the most common mutation c.191dupA; 3 patients. Sequence capture – targeted resequencing.")
33
Sequence Capture and Targeted Resequencing, SeqCap-TR
Fenotypové projevy neuromuskulárních chorob mohou být společné pro nemoci asociované s mutacemi v různých genech a často nelze rozlišit, o jaký konkrétní typ svalové dystrofie se jedná. Sequence capture-targeted resequencing Vybrané nemoci (geny): Duchennova svalová dystrofie (gen DMD); Emery-Dreifussova svalová dystrofie (geny EMD, FHL1, LMNA); pletencové svalové dystrofie, dominantní (geny MYOT, LMNA, CAV3); pletencové svalové dystrofie, recesivní (geny CAPN3, DYSF, SGCG, SGCA, SGCB, SGCD, TCAP, TRIM32, FKRP, TTN, POMT1, ANO5, FKTN, POMT2, POMGNT1); kongenitální svalové dystrofie (LAMA2, LARGE, SEPN1, COL6A1, COL6A2, COL6A3, ITGA7, DNM2); kongenitáoní myopatie, distální myopatie, and další myopatie (NEB, TPM3, ACTA1, TPM2, TNNT1, CFL2, RYR, MTM1, BIN, CRYAB, DES, LAMP2, PABPN1). Analýza exonů a přilehlých intronových oblastí 42 genů, pb.
: Duchennova svalová dystrofie (gen DMD); Emery-Dreifussova svalová dystrofie (geny EMD, FHL1, LMNA); pletencové svalové dystrofie, dominantní (geny MYOT, LMNA, CAV3); pletencové svalové dystrofie, recesivní (geny CAPN3, DYSF, SGCG, SGCA, SGCB, SGCD, TCAP, TRIM32, FKRP, TTN, POMT1, ANO5, FKTN, POMT2, POMGNT1); kongenitální svalové dystrofie (LAMA2, LARGE, SEPN1, COL6A1, COL6A2, COL6A3, ITGA7, DNM2); kongenitáoní myopatie, distální myopatie, and další myopatie (NEB, TPM3, ACTA1, TPM2, TNNT1, CFL2, RYR, MTM1, BIN, CRYAB, DES, LAMP2, PABPN1). Analýza exonů a přilehlých intronových oblastí 42 genů, pb.")
Podobné prezentace




 B. Ravčuková , J>")














