Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Bioinformatika Jiří Vondrášek Jan Pačes http://bio.img.cas.cz/kurs
Ústav organické chemie a biochemie Jan Pačes Ústav molekulární genetiky Úvodní stránka

2
Predikce sekundární struktury
v proteinech

15
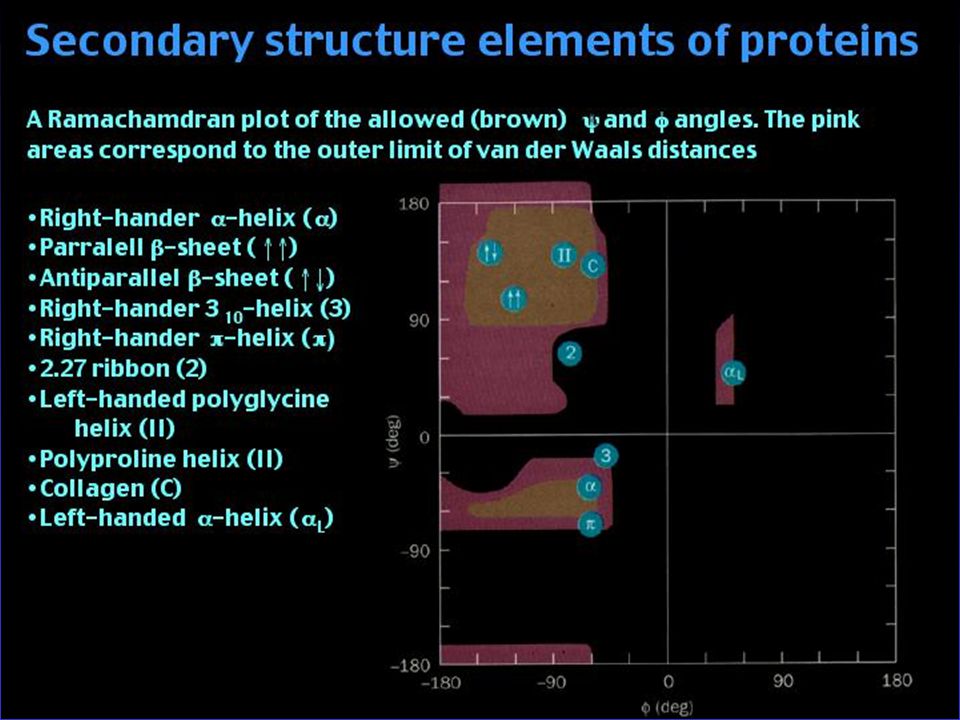
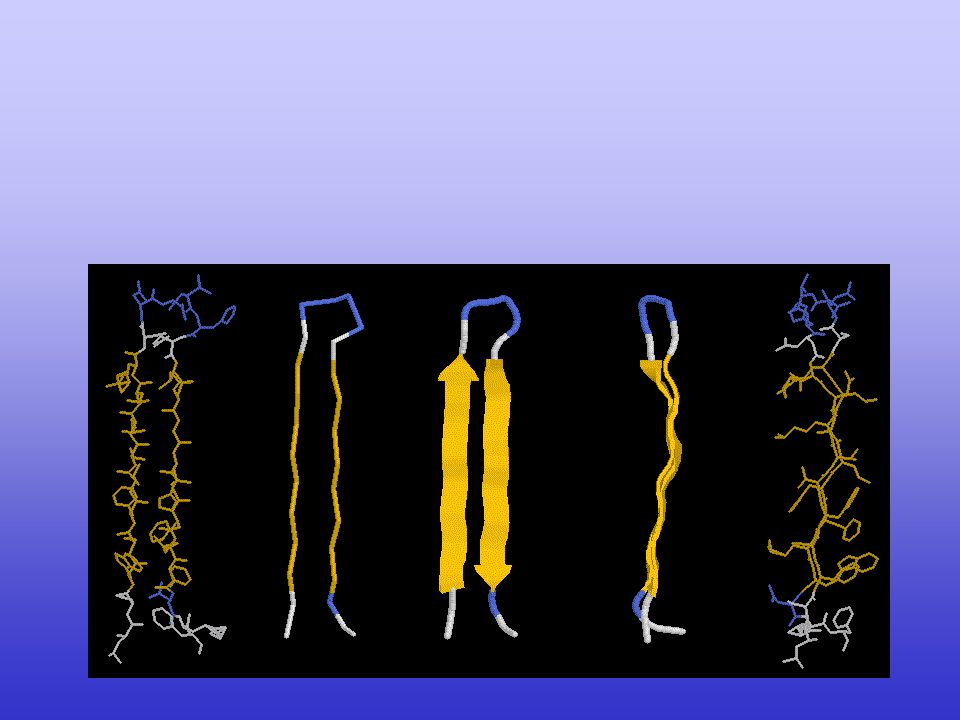
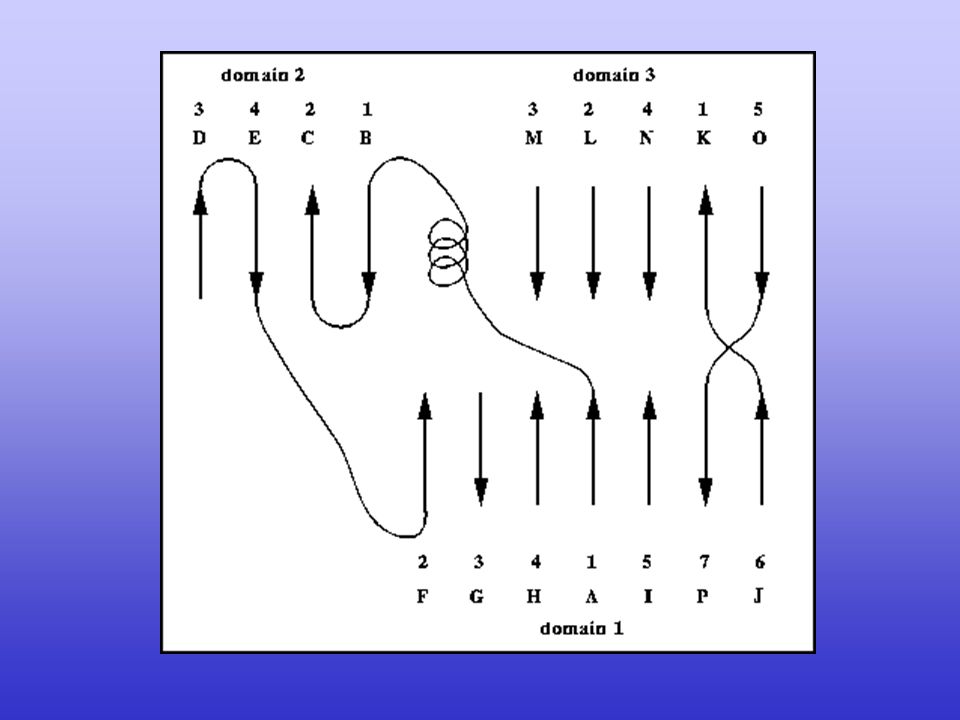
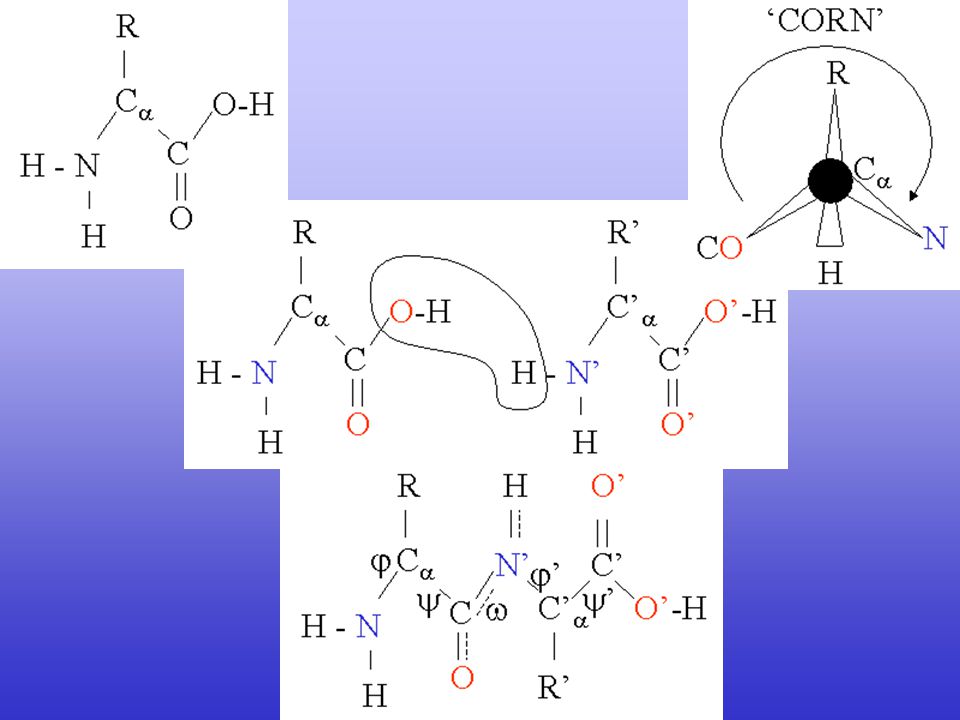
Secondary structure Elements in Protein

16
B Sheets – atom representations

21
a helices - atom representations

24
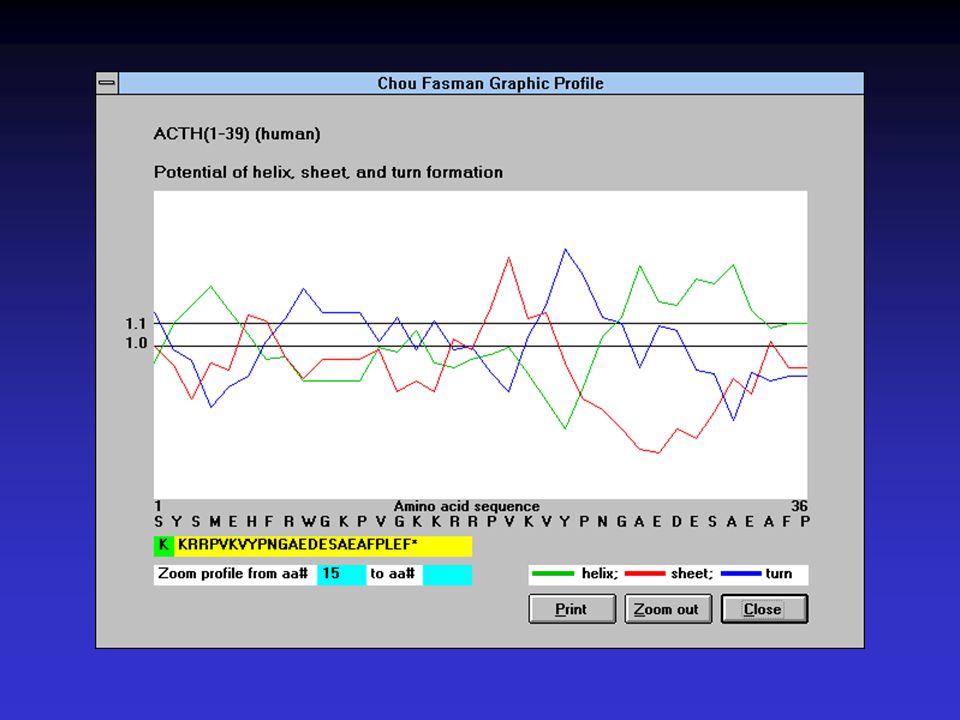
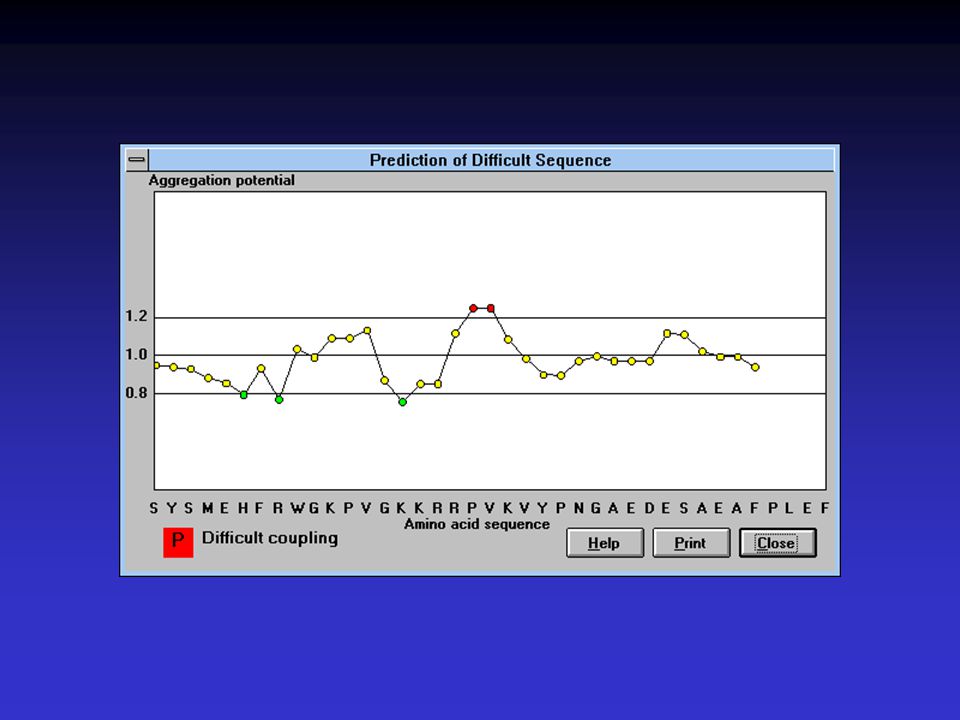
Metoda Chou-Fasman Využívá tabulky konformačních parametrů
extrahovaných z reálných struktur a CD spektroskopie Tabulka obsahuje pravděpodobnosti pro jednotlivé sekundární prvky pro každou aminokyselinu

25
Assign all of the residues in the peptide the appropriate set of parameters.
Scan through the peptide and identify regions where 4 out of 6 contiguous residues have P(a-helix) > 100. That region is declared an alpha-helix. Extend the helix in both directions until a set of four contiguous residues that have an average P(a-helix) < 100 is reached. That is declared the end of the helix. If the segment defined by this procedure is longer than 5 residues and the average P(a-helix) > P(b-sheet) for that segment, the segment can be assigned as a helix. Repeat this procedure to locate all of the helical regions in the sequence.
 > 100. That region is declared an alpha-helix. Extend the helix in both directions until a set of four contiguous residues that have an average P(a-helix) < 100 is reached. That is declared the end of the helix. If the segment defined by this procedure is longer than 5 residues and the average P(a-helix) > P(b-sheet) for that segment, the segment can be assigned as a helix. Repeat this procedure to locate all of the helical regions in the sequence.")
26
3. Scan through the peptide and identify a region where 3 out of 5 of the residues have a value of P(b-sheet) > 100. That region is declared as a beta-sheet. Extend the sheet in both directions until a set of four contiguous residues that have an average P(b-sheet) < 100 is reached. That is declared the end of the beta-sheet. Any segment of the region located by this procedure is assigned as a beta-sheet if the average P(b-sheet) > 105 and the average P(b-sheet) > P(a-helix) for that region. 4. Any region containing overlapping alpha-helical and beta-sheet assignments are taken to be helical if the average P(a-helix) > P(b-sheet) for that region. It is a beta sheet if the average P(b-sheet) > P(a-helix) for that region.
 > P(b-sheet) for that region. It is a beta sheet if the average P(b-sheet) > P(a-helix) for that region.")
27
5. To identify a bend at residue number j, calculate the following value p(t) = f(j)f(j+1)f(j+2)f(j+3) 6. where the f(j+1) value for the j+1 residue is used, the f(j+2) value for the j+2 residue is used and the f(j+3) value for the j+3 residue is used. If: (1) p(t) > ; (2) the average value for P(turn) > 1.00 in the tetrapeptide; and (3) the averages for the tetrapeptide obey the inequality P(a-helix) < P(turn) > P(b-sheet), then a beta-turn is predicted at that location.
 value for the j+1 residue is used, the f(j+2) value for the j+2 residue is used and the f(j+3) value for the j+3 residue is used. If: (1) p(t) > ; (2) the average value for P(turn) > 1.00 in the tetrapeptide; and (3) the averages for the tetrapeptide obey the inequality P(a-helix) < P(turn) > P(b-sheet), then a beta-turn is predicted at that location.")
28
CHOU-FASMAN RULES FOR ALPHA HELIX:
Helical residues = >1.0 for helix. Helical breakers = 4/6 >1.0 nucleates helix. Helix continues both ways until 4 contiguous Special rules for Proline. Segment 5 residues or longer and P(a) > P(b) = helix. CHOU-FASMAN RULES FOR BETA STRAND: Beta residues = >1.0 for strand. Beta breakers = 3/5 >1.0 nucleates strand. Strand continues both ways until 4 contiguous Segment with average P(b) > 1.05 and P(b) > P(a) = strand
 > P(b) = helix. CHOU-FASMAN RULES FOR BETA STRAND: Beta residues = >1.0 for strand. Beta breakers = 3/5 >1.0 nucleates strand. Strand continues both ways until 4 contiguous. Segment with average P(b) > 1.05 and P(b) > P(a) = strand.")
29
Name P(a) P(b) P(turn)f(i) f(i+1) f(i+2) f(i+3)
Alanine Arginine Aspartic Acid Asparagine Cysteine Glutamic Acid Glutamine Glycine Histidine Isoleucine Leucine Lysine Methionine Phenylalanine Proline Serine Threonine Tryptophan Tyrosine Valine

30
Chou-Fasman propensities (partial table)
")
33
Garnier-Osguthorpe-Robson GOR
Využívá tabulku tendencí určenou primárně z krystalových struktur Tabulka obsahuje jednu pravděpodobnost pro každou strukturu a každou aminokyselinu v okně dlouhém 17 aminokyselin

34
Teorie informace aplikovaná na predikci struktury
Jakou informaci získáme o pravděpodobnosti, že residuum j je v jistém stavu (H,E,T,C), ze znalosti jaké residuum je v pozici jm (m £ 8), nezávisle na tom co je residuum j zač. Je li m=0, podobné Chou-Fasman
, ze znalosti. jaké residuum je v pozici jm (m £ 8), nezávisle. na tom co je residuum j zač. Je li m=0, podobné Chou-Fasman.")
35
pro H

36
Přesnost predikce Obě metody mají přesnost kolem 55 – 65%
Hlavní důvod je to, že přeceňují lokální kontext na úkor globálního, tedy typ proteinu Tytéž aminokyseliny mohou zaujímat různé konfigurace v cytoplazmatickém a v membránovém proteinu

42
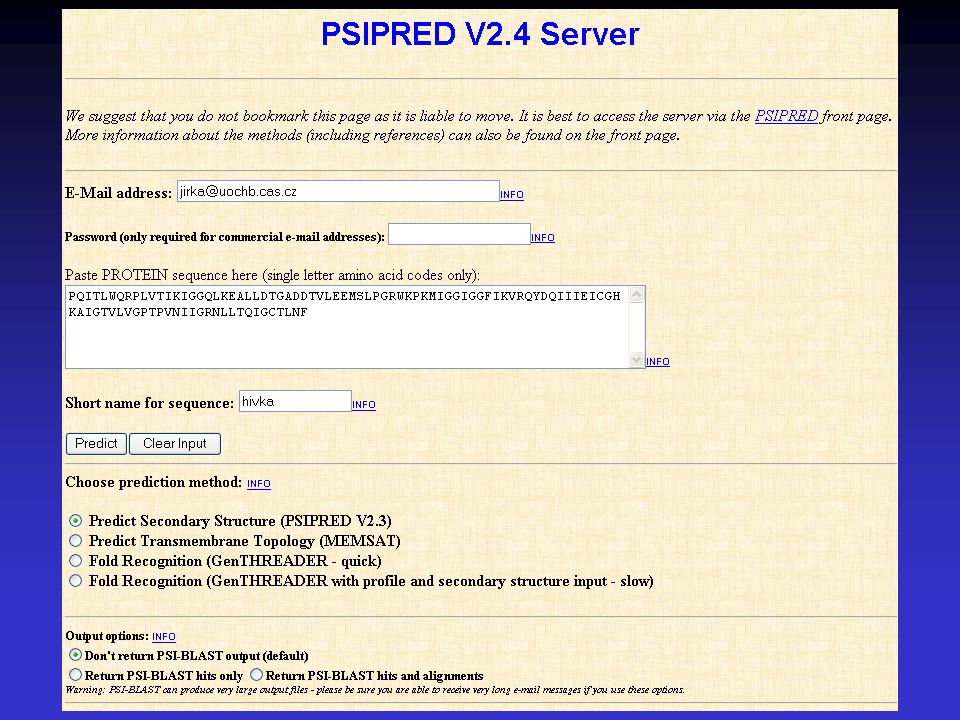
PSIPred output window

54
PSIPRED PREDICTION RESULTS
Key Conf: Confidence (0=low, 9=high) Pred: Predicted secondary structure (H=helix, E=strand, C=coil) AA: Target sequence Conf: Pred: CCCCEECCCEEEEEECCHHHHHHHHCCCCCHHHHHCCCCCCCCCCEEECCCCEEEEEEEC AA: PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD Conf: Pred: CEEEEECCCCCEEEEEECCCCHHHHHHHHHHHHCCCCCC AA: QIIIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF Calculate PostScript, PDF and JPEG graphical output for this result using:
 Pred: Predicted secondary structure (H=helix, E=strand, C=coil) AA: Target sequence. Conf: Pred: CCCCEECCCEEEEEECCHHHHHHHHCCCCCHHHHHCCCCCCCCCCEEECCCCEEEEEEEC. AA: PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYD Conf: Pred: CEEEEECCCCCEEEEEECCCCHHHHHHHHHHHHCCCCCC. AA: QIIIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF Calculate PostScript, PDF and JPEG graphical output for this result using: id=")
61
Sekundární strukturní prvky – formulace problému
• Daná proteinová sekvence – NWVLSTAADMQGVVTDGMASGLDKD... • Predikce sekvence sekundární struktury: – LLEEEELLLLHHHHHHHHHHLHHHL... • „3-state“ problém: {ARNDCQEGHILKMFPSTWYV}n-> {L,H,E}n

62
Predikce prvků sekundární struktury u proteinů
motivace pro předpověď prvků sekundární struktury - efektivní konformační vzorek pro 3D protein folding - vylepšení ostatních sekvenčních a strukturně analytických metod : sekvenční alignment : homologické a „threading“ modelování (CASP) : analýza experimentálních dat : protein design
 : analýza experimentálních dat. : protein design.")
63
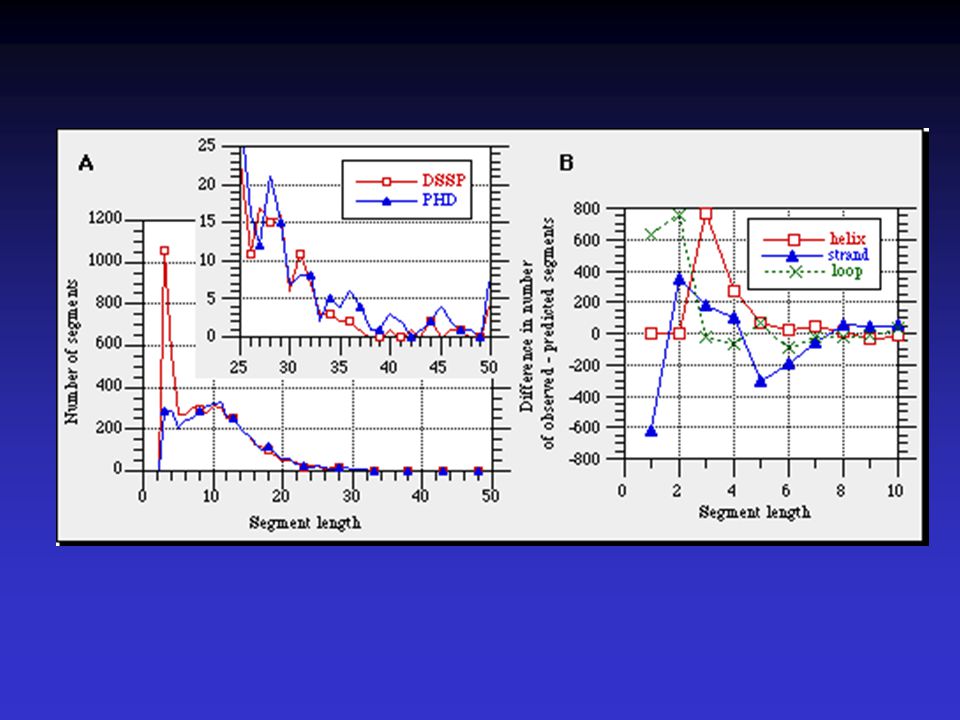
V proteinech se známou strukturou není určení sekundární
struktury jednoznačným a jednoduchým úkolem Dva základní klasifikační proramy a postupy pro určení SS z krystalových struktur DSSP a STRIDE výsledky těchto dvou postupů se liší nepatrně Reference Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers Dec;22(12):
:")
64
Výskyt aminokyselin a jejich distribuce v
příslušných prvcích sekundární struktury by měly být vodítkem při predikci prvků SS stupeň determinace Klasické metody Chou Fasman GOR (Garnier-Osguthorpe-Robson)
")
65
Adaptivní metody Metoda neuronových sítí
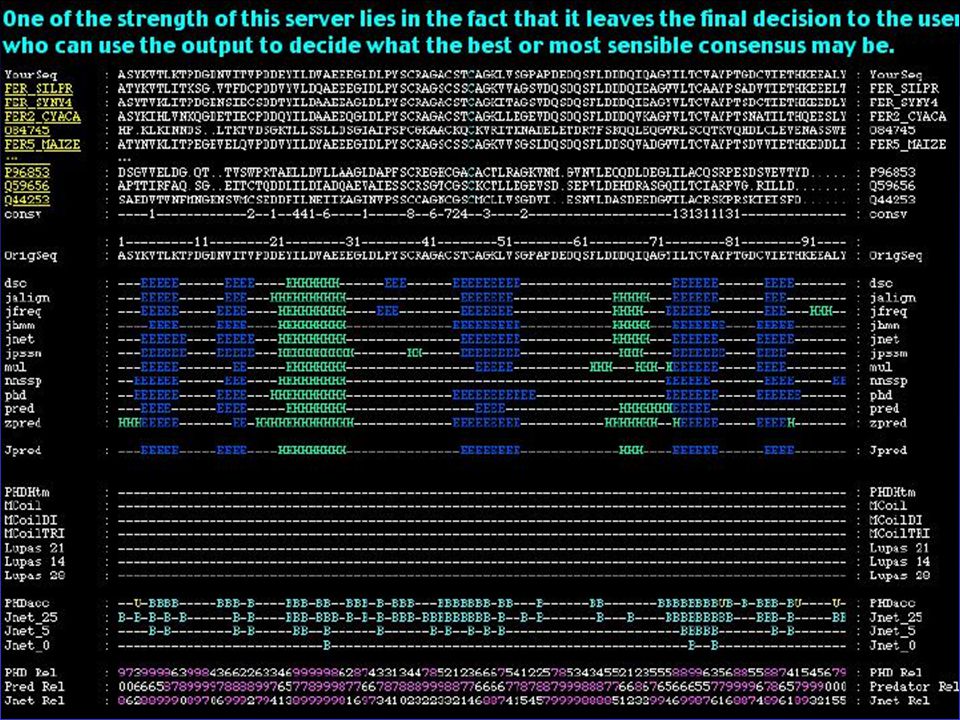
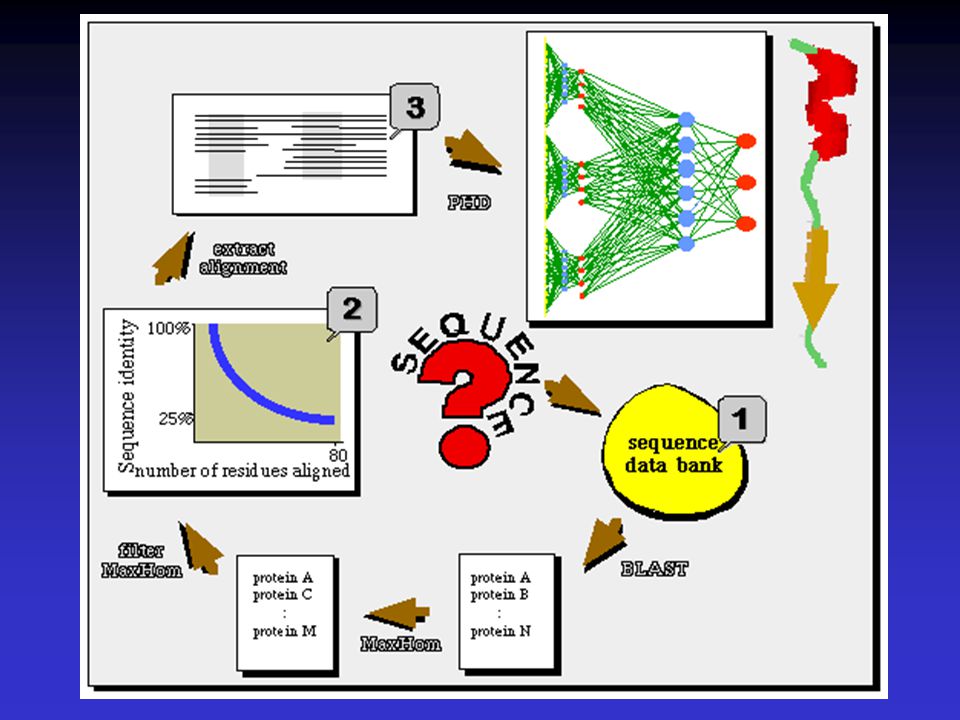
pokusná síť používá sadu známých proteinů k predikci žádané struktury ze sekvenčních dat <nnpredict> -Metoda založená na homologii hledané sekvence se známými proteiny <SOPM> <PHD>

66
Neural Network methods
A neural network with multiple layers is presented with known sequences and structures - network is trained until it can predict those structures given those sequences Allows network to adapt as needed (it can consider neighboring residues like GOR)
")
Podobné prezentace












































>")




, Ω(n 2 ), Θ(n·log 2 (n)), … Různé algoritmy mají různou složitost: O(n), Ω(n 2 ), Θ(n·log.>")
