Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Enzymopatie – dědičné poruchy metabolismu RNDr
Enzymopatie – dědičné poruchy metabolismu RNDr. Hana Zoubková, PhD Energie - 228

3
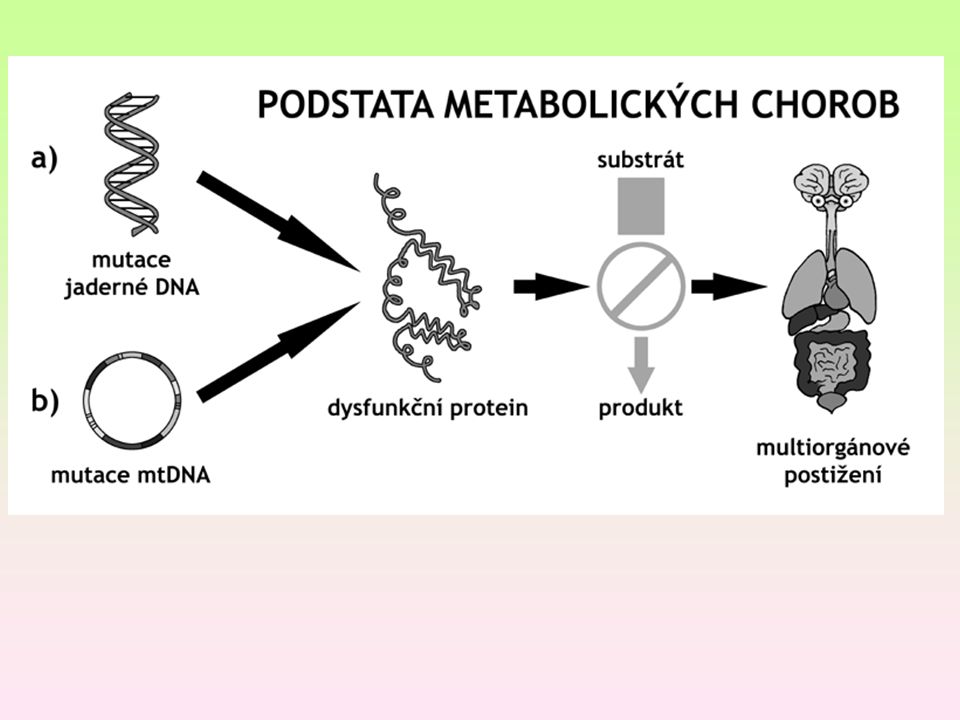
Obecné vlastnosti enzymopatií
Změny enzymů zapříčiňující vrozené poruchy metabolismu. Jsou téměř vždy recesivní. difúzní versus makromolekulární substráty Poruchy dány: akumulací substrátu (1), deficitem produktu (2) nebo vznikem látek, které jsou pro tělo toxické (3) nebo kombinace – heterozygoti s 50% reziduální aktivitou jsou klinicky normální Difúzní – deriváty se pohybují tělem a poškozují i buňky, které nemají k poškozenému enzymu žádný vztah Makromolekulární omezeny na tkáně, kde se makromolekula akumuluje
, deficitem produktu (2) nebo vznikem látek, které jsou pro tělo toxické (3) nebo kombinace. – heterozygoti s 50% reziduální aktivitou jsou klinicky normální. Difúzní – deriváty se pohybují tělem a poškozují i buňky, které nemají k poškozenému enzymu žádný vztah. Makromolekulární omezeny na tkáně, kde se makromolekula akumuluje.")
4
Obecné vlastnosti enzymopatií
Ad 1 - hromadění molekul, bílkoviny, enzymu, které nejsou správným způsobem odbourány a vyloučeny Ad 2. nepřítomnost molekuly bílkoviny, enzymu nebo jiné látky v organismu Ad 3. vznik látek (bílkovin, enzymů nebo jiných látek), které do organismu nepatří, a způsobují jeho akutní nebo pozvolnou otravu
, které do organismu nepatří, a způsobují jeho akutní nebo pozvolnou otravu.")
5
Obecné vlastnosti enzymopatií
Jediný pacient může mít ztrátu funkce více než jednoho enzymu. Existue fenotypová homologie pro enzymové defekty. Netýká se několika katalytických ribonukleových kyselin. Používají stejný kofaktor, sdílejí společný aktivační, modifikující nebo stabilizující protein, může chybět celá skupina enzymů nebo je abnormální celá organela Nemoci způsobené jinými enzymy ale ve stejné oblasti metabolismu, nebo různé nemoci, které vznikají v důsledku parciálního nebo kompletního defektu enzymu

6
Obecné vlastnosti enzymopatií
V současné době se rozeznává více než 850 DPM, z nichž léčitelných nebo ovlivnitelných dietou je přibližně 100. nízkobílkovinná, bezláktózová, belepková dieta Při současné porodnosti v České republice každým rokem se narodí kolem dětí s DPM. Většina praktických lékařů pro děti a dorost se s nimi nesetká.

7
Organické acidurie (acidemie)
Klasifikace DPM AMK – hyperfenylalaninemie, Tyrosinémie, Alkaptonúrie, Homocystinurie, Cystinurie, Cystinóza, choroba javorového sirupu nebo-li Leucinóza Organické acidurie (acidemie) DPM sacharidů – Galaktosémie, deficit galaktokinázy, deficit uridindifosfát 4-epimerázy, intolerance fruktózy, deficit fructosa-1,6-bifosfatásy, glykogenosy DPM purinů a pyrimidinů – Porfyrie DPM lysozomů: Tay-Sachsova porucha DPM komplexních molekul : lysozomy, peroxisomy, DMP mitochondrií (metylmalonová acidurie, propionová acidurie, izovalerová acidurie) Poruchy cyklu močoviny
 DPM sacharidů – Galaktosémie, deficit galaktokinázy, deficit uridindifosfát 4-epimerázy, intolerance fruktózy, deficit fructosa-1,6-bifosfatásy, glykogenosy. DPM purinů a pyrimidinů – Porfyrie. DPM lysozomů: Tay-Sachsova porucha. DPM komplexních molekul : lysozomy, peroxisomy, DMP mitochondrií. (metylmalonová acidurie, propionová acidurie, izovalerová acidurie) Poruchy cyklu močoviny.")
8
Fenylalanin

9
Hyperfenylalaninémie
Příčinou jsou ztrátové mutace v genu pro enzym fenylalanin hydroxyláza (PAH) nebo v metabolismu jejího kofaktoru tetrahydrobiopterinu. = skupina onemocnění: Fenylketonúrie PKU Variantní PKU Nefenylketonurická hyperfenylalaninémie Non-PKU Ztrátové mutace jsou mutace vedoucí ke ztrátě funkce proteinu. Nefenylketonurická hyperfenylalaninémie Reziduální aktivita fenylalanin hydroxylázy, plazm koncentrace fenylalaninu je pod 1mM, což je10x zvýšený oproti normálu a nižší než u klasické PKU Variantní PKU přechod mezi non-PKU a PKU 9
 nebo v metabolismu jejího kofaktoru tetrahydrobiopterinu. = skupina onemocnění: Fenylketonúrie PKU. Variantní PKU. Nefenylketonurická hyperfenylalaninémie Non-PKU. Ztrátové mutace jsou mutace vedoucí ke ztrátě funkce proteinu. Nefenylketonurická hyperfenylalaninémie. Reziduální aktivita fenylalanin hydroxylázy, plazm koncentrace fenylalaninu je pod 1mM, což je10x zvýšený oproti normálu a nižší než u klasické PKU. Variantní PKU přechod mezi non-PKU a PKU. 9.")
10
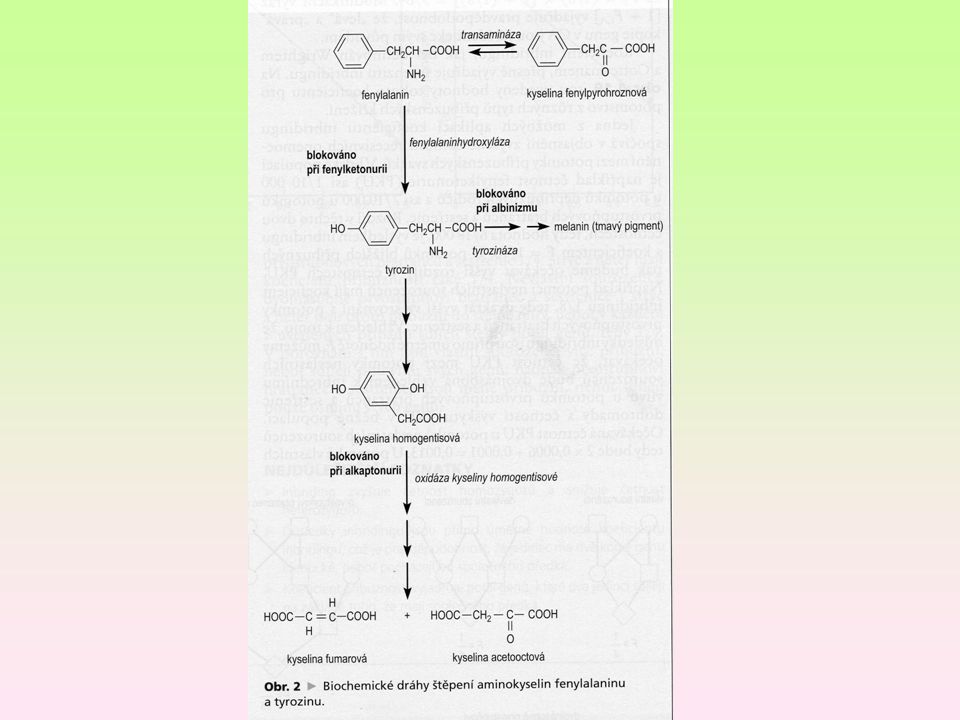
Fenylketonúrie – PKÚ Dána neschopností metabolizovat aromatickou aminokyselinu fenylalanin na tyrozin, ta se změní na kyselinu fenylpyrohroznovou, což je toxický vedlejší produkt, který je příčinou mentální retardace, světlá pigmentace. chromozom 12q24.1, incidence 1:5 000 a 1: živých narozených dětí, 1:6 000 v ČR, ročně je diagnostikováno okolo 10 případů vysoká koncentrace fenylpyruvátu v tkáních a tekutinách, poškozuje myelinizaci nerv. vláken, při synthéze mastných kyselin a cholesterolu -- nedostatek myelinových obalů V bílkovinách rostlinného i živočišného původu, z tohoto důvodů je třeba vynechat maso, mléko, obilniny, běžné pečivo, cereálie, většinu cukrovinek, ořechy, luštěniny, částečně i většinu druhů ovoce a zeleniny a potraviny obsahující umělé sladidlo aspartam. 10

11
maso, mléko, obilniny, běžné pečivo, cereálie, většinu cukrovinek, ořechy, luštěniny, částečně ovoce a zeleninu a potraviny obsahující umělé sladidlo aspartam

12
Alelická i lokusová heterogenita
400 mutovaných alel v každém ze 13 exonů mutace, 6 mutací u 2/3 evropské populace, další 6 mutací 80% asijské populace Novorozenecký screening Test je lépe neprovádět dříve než třetí či čtvrtý den věku dítěte. Léčba PKÚ - úprava stravy – je účinná, je-li zahájena hned po narození Gen pro PAH izolován v 1986 Alelová heterogenita – 400 alel, většinou vzácné, v každém ze 13 exonů mutace 6 mutací – složení heterozygoti 2/3 evropské populace, další 6 mutací 80% asijské populace, nov scree PKU, congenita hypothyroidism, galactosemia, sickle cell disease, cystic fibrosis,

14
Tyrosin

15
Albinismus klasický, typ IA.
chybění pigmentu kůže a vlasů, nedostatek produktu příčinou je nedostatek, nefukčnost tyrozinázy Typ IA - tyrozináza negativní Typ IB – tyrozináza je pozitivní, neaktivní Type II – nejrozsáhlejší, tyrozináza pozitivní, ale s mutací genu Enzym v syntéze proteinů, katecholaminů, melaninu a tyroidního hormonu

16
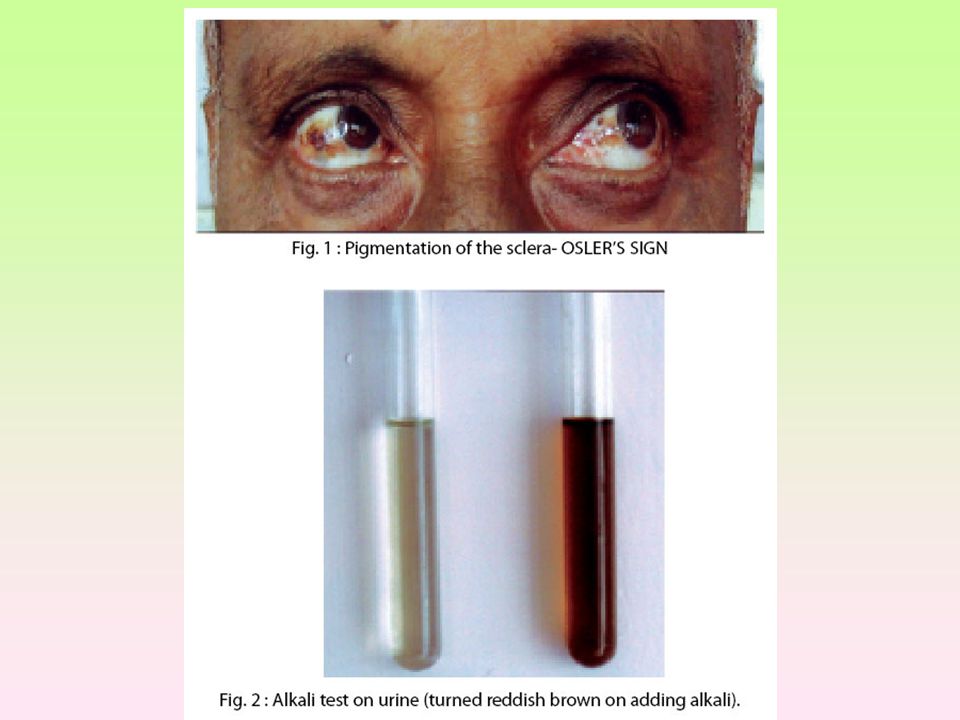
Alkaptonúrie - AKU Je dána neschopností metabolizovat kyselinu homogentisovou, příčinou je nedostatek enzymu homogentizát dioxygenázy (HGD), posléze se hromadí substrát hnědo-černý pigment - alkapton v moči Ochronóza – pigmentace pojivových tkání Incidence 1: Homogentizát je následne oxidovaný vzdušným kyslíkom na alkapton Léčba: omezený přísun fenyalaninu a tyrosinu
, posléze se hromadí substrát. hnědo-černý pigment - alkapton v moči. Ochronóza – pigmentace pojivových tkání. Incidence 1: Homogentizát je následne oxidovaný vzdušným kyslíkom na alkapton. Léčba: omezený přísun fenyalaninu a tyrosinu.")
18
Tyrosin

19
Tyrosinémie typ I Je porucha metabolismu tyrosinu, porucha aktivity fumarylacetoacetát hydrolázy (FAH) hromadí se tyrosin (substrát) i toxické vedlejší metabolity, nejzávažnější 1: (jeden případ za jeden až dva roky) Tyrosinémie typ II a III jsou vzácnější Hladina toxického metabolitu sukcinylacetonu není tak vysoká. Léčba alkaptonurie a tyrosinemie dietou a Nitisonem. zejména sukcinylacetonu klinické příznaky tyrosinémie typu I jsou velmi různorodé s postižením jaterních buněk a ledvinných tubulů
 i toxické vedlejší metabolity, nejzávažnější. 1: (jeden případ za jeden až dva roky) Tyrosinémie typ II a III jsou vzácnější. Hladina toxického metabolitu sukcinylacetonu není tak vysoká. Léčba alkaptonurie a tyrosinemie dietou a Nitisonem. zejména sukcinylacetonu. klinické příznaky tyrosinémie typu I jsou velmi různorodé s postižením jaterních buněk a ledvinných tubulů.")
21
Metionin Cystein

22
Odbourávání metioninu

23
Homocystinurie - sirná AMK
Je dána nedostatkem cystationin β-syntázy (CBS), což způsobuje zvýšenou hodnotu homocysteinu a metioninu v moči, dochází k hromadění substrátu. Metionin není metabolizován přes homocystein na cystein. Postiženy čtyři orgánové systémy: oko, skelet (kost), cévní endotel (cévní výstelka) a centrální nervový systém Esenciální aminokyselina se sírou – přítomnost v protein. syntéze je kritická, v syntéze karnitinu, taurinu, fosphatidylcholinu, fosfolipidu………..heparinu, koenzym A, biotinu, glutationu 132 mutací, většina je missense mutace
, což způsobuje zvýšenou hodnotu homocysteinu a metioninu v moči, dochází k hromadění substrátu. Metionin není metabolizován přes homocystein na cystein. Postiženy čtyři orgánové systémy: oko, skelet (kost), cévní endotel (cévní výstelka) a centrální nervový systém. Esenciální aminokyselina se sírou – přítomnost v protein. syntéze je kritická, v syntéze karnitinu, taurinu, fosphatidylcholinu, fosfolipidu………..heparinu, koenzym A, biotinu, glutationu. 132 mutací, většina je missense mutace.")
24
Ectopia lensis, nadměrná výška, délka končetin, vaskulární abnormality

25
Valin Leucin Isoleucin

26
Organické acidurie (acidemie)
Skupina poruch specifických enzymů v katabolismu větvených aminokyselin leucinu, valinu a isoleucinu. Jsou dány hromaděním toxických organických kyselin (vedlejších produktů). Nastává překyselení organismu, postižen je hlavně mozek. Každým rokem se narodí v České republice do 10 pacientů s některou z organických acidurií.
. Nastává překyselení organismu, postižen je hlavně mozek. Každým rokem se narodí v České republice do 10 pacientů s některou z organických acidurií.")
27
„Maple syrup urine disease - MSUD“
nebo-li Leucinóza Je to porucha met. větvených α-ketokyselin, porucha příslušné dehydrogenázy v multienzymovém komplexu. Hromadí se keto - kyseliny v těle, způsobují zápach moči a ušního mazu po javorovém sirupu. Incidence 1: – , Mennonité – 1:1000 multienzymový komplex volně asociovaný s vnitřními membránami mitochondrie keto - kyseliny v těle, způsobují nesnášenlivost potravy, neprospívání, zvracení, letargie a zápach moči a ušního mazu po javorovém sirupu

28
Galaktóza

29
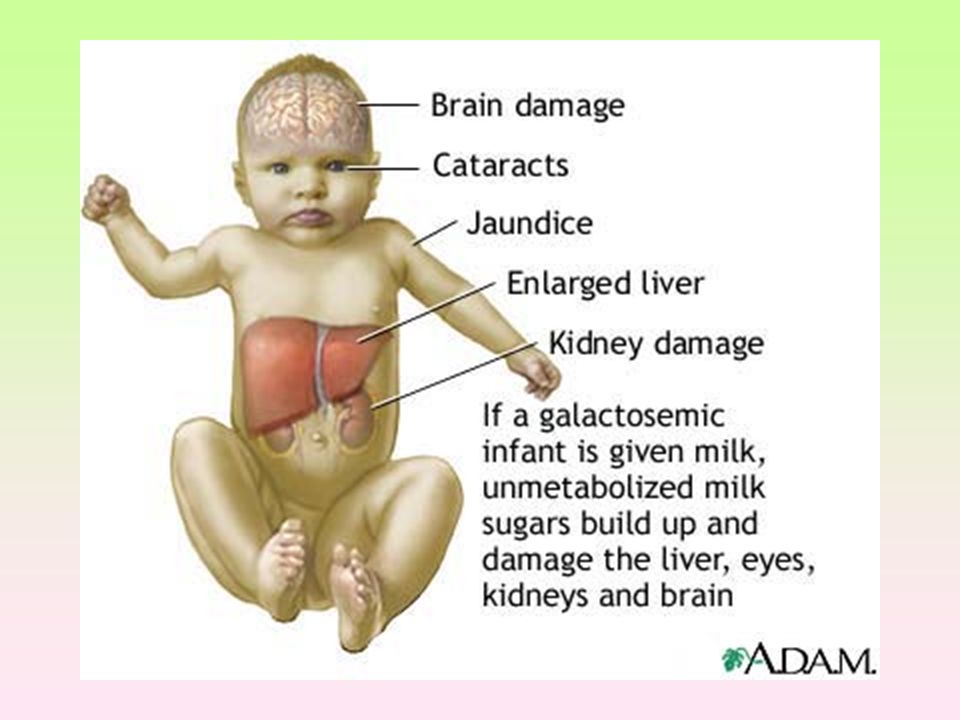
Galaktosémie, klasická
Neschopnost metabolizovat monosacharid galaktózu (komponentu laktózy), vznikají toxické vedlejší produkty metabolismu, které působí toxicky na játra, mozek, ledviny a oční čočky. Je dána nefunkčností Galaktózo-1-fosfát uridyl-transferázy – GALT (9.chromozom, 160 mutací) 1: až 1:70 000 V cerebrosidech, gangliosidech, glykoproteinech hromadí galaktóza-1-P, která se dále metabolizuje alternativní cestou na galaktitol, Galakto-1-fosfát na glukozo-1-fosfát, způsobují podvýživu, cirhózu jater, kataraktu, mentální retardaci Určité množství galaktózy je přítomno ve všech druzích ovoce a v některých obilovinách, zelenině (například kapusta, květák, řepa, růžičková kapusta, zelí, rajčata) a luštěninách (hrách, sója, fazole, čočka apod.). U pacientů je nutno vyloučit z jídelníčku kakao, čokoládu, smetanové zmrzliny, mandle, ořechy, skořici.
, vznikají. toxické vedlejší produkty metabolismu, které působí toxicky na játra, mozek, ledviny a oční čočky. Je dána nefunkčností Galaktózo-1-fosfát uridyl-transferázy – GALT (9.chromozom, 160 mutací) 1: až 1: V cerebrosidech, gangliosidech, glykoproteinech. hromadí galaktóza-1-P, která se dále metabolizuje alternativní cestou na galaktitol, Galakto-1-fosfát na glukozo-1-fosfát, způsobují podvýživu, cirhózu jater, kataraktu, mentální retardaci. Určité množství galaktózy je přítomno ve všech druzích ovoce a v některých obilovinách, zelenině (například kapusta, květák, řepa, růžičková kapusta, zelí, rajčata) a luštěninách (hrách, sója, fazole, čočka apod.). U pacientů je nutno vyloučit z jídelníčku kakao, čokoládu, smetanové zmrzliny, mandle, ořechy, skořici.")
31
Tay-Sachsova porucha GM2-gangliozidóza, je dána neschopností degradovat sfingolipid GM2-gangliosid, dána defektem hexozaminidázy A (HEX A). Nastává hromadění substrátu, hlavně se vyskytuje těžká infantilní forma. Vyskytuje se třešňovo-červená skvrna na sítnici oka Zvýšený výskyt (1:3600) u aškenázské židovské populace (1: , chromozom 15) Lysozomální střádací choroba, postižen hlavně mozek, neurodegenerace Úmrtí mezi 2. až 4. rokem života lipidy obsahující dlouhý řetězec s bazickou skupinou; jejich základem je ceramid, sloučenina obsahující alkohol sfingosin. Podle další látky, která je vázána na ceramid, se s. dále dělí na sfingomyeliny (obsahují fosforylcholin) a sfingoglykolipidy (obsahují molekuly cukrů). Tato skupina zahrnuje cerebrosidy (obsahují molekulu glukosy nebo galaktosy), sulfatidy (cerebrosidy, které však dále obsahují zbytek kyseliny sírové) a gangliosidy (obsahují neuraminovou kyselinu).
. Nastává hromadění substrátu, hlavně se vyskytuje těžká infantilní forma. Vyskytuje se třešňovo-červená skvrna na sítnici oka. Zvýšený výskyt (1:3600) u aškenázské židovské populace (1: , chromozom 15) Lysozomální střádací choroba, postižen hlavně mozek, neurodegenerace. Úmrtí mezi 2. až 4. rokem života. lipidy obsahující dlouhý řetězec s bazickou skupinou; jejich základem je ceramid, sloučenina obsahující alkohol sfingosin. Podle další látky, která je vázána na ceramid, se s. dále dělí na sfingomyeliny (obsahují fosforylcholin) a sfingoglykolipidy (obsahují molekuly cukrů). Tato skupina zahrnuje cerebrosidy (obsahují molekulu glukosy nebo galaktosy), sulfatidy (cerebrosidy, které však dále obsahují zbytek kyseliny sírové) a gangliosidy (obsahují neuraminovou kyselinu).")
33
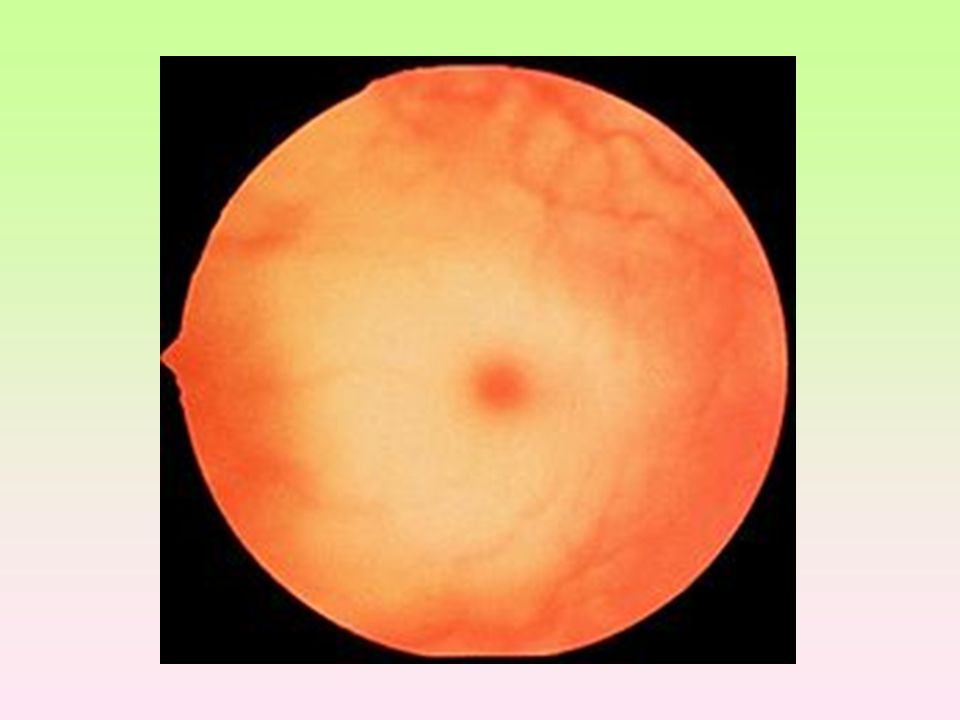
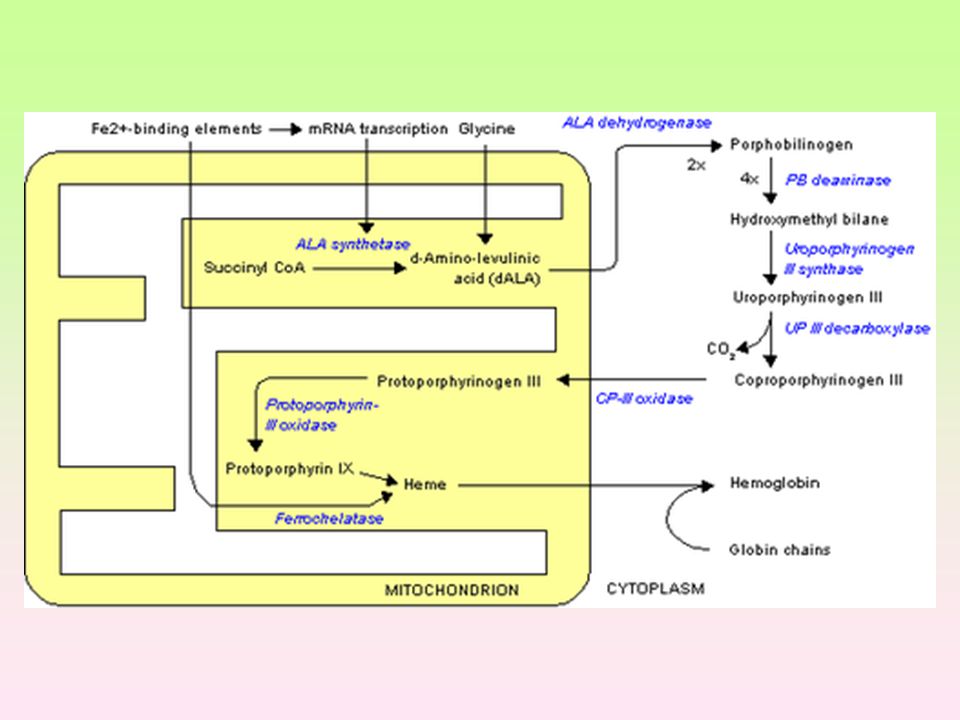
Porfyrie Dána mutací genů, které řídí syntézu enzymů působících při syntéze hemu. Defekty v časné fázi způsobují hromadění výchozích látek: kyselina 5-aminolevulová a porfobilinogen - důsledek je hromadění substrátu Defekty ve vyšších stupních syntézy vedou k hromadění porfyrinogenů, nastává hromadění substrátu a vedlejších produktů kožní projevy, jaterní Vrozená (kongenitální) erytropoetická porfyrie (Güntherova choroba) Hromadění substrátu s toxickým účinkem – neuropatie (svalová slabost), abdominální bolest, neklid, hysterie, psychotické stavy. (reakci na viditelné světlo v oblasti 400 nm); při vystavení porfyrinů světlu o této vlnové délce dochází k jejich excitaci. Porfyrinogeny, jejichž oxidační produkty způsobují fotosenzitivitu reakci s molekulárním kyslíkem za tvorby kyslíkových radikálů, které poškozují buněčné organely včetně lysosomů → dochází k uvolnění lysosomálních enzymů, které poškozují světlu vystavenou kůži (erytém, puchýře, jizvení). Akutními porfyriemi jsou častěji postiženy ženy, chronickými muži. Nejvíce postižených orgánů produkujících porfyriny Hepatální, erytropoetické, erytrohepatální
 erytropoetická porfyrie (Güntherova choroba) Hromadění substrátu s toxickým účinkem – neuropatie (svalová slabost), abdominální bolest, neklid, hysterie, psychotické stavy. (reakci na viditelné světlo v oblasti 400 nm); při vystavení porfyrinů světlu o této vlnové délce dochází k jejich excitaci. Porfyrinogeny, jejichž oxidační produkty způsobují fotosenzitivitu reakci s molekulárním kyslíkem za tvorby kyslíkových radikálů, které poškozují buněčné organely včetně lysosomů → dochází k uvolnění lysosomálních enzymů, které poškozují světlu vystavenou kůži (erytém, puchýře, jizvení). Akutními porfyriemi jsou častěji postiženy ženy, chronickými muži. Nejvíce postižených orgánů produkujících porfyriny Hepatální, erytropoetické, erytrohepatální.")
35
Srovnání jaderné a mitochondriální dědičnosti
Mutované geny jaderné Mutované geny mitochondriální Kombinace gen. informace Genetická informace od matky otce a matky Mendelovy zákony Maternální dědičnost Postižení celého těla Variabilní exprese

36
- 2 ribosomální RNA, 22 transferové RNA, 13 polypeptides
- enzymes oxidative phosphorylation (ATP): cytochrom c oxidase, cytochrom b, components of ATPase complex, components NADH dehydrogenase and NADH reductase
: cytochrom c oxidase, cytochrom b, components of ATPase complex, components NADH dehydrogenase and NADH reductase.")
37
Mitochondriální choroby
heterogenní skupina chorob s dysfunkcí dýchacího řetězce, jsou to metabolické poruchy a neurodegenerativní či svalové choroby z nedostatek produktu. Tkáně s vysokou spotřebou ATP (mozek, svaly, játra, srdce, ledviny, endokrinní žlázy) jsou postiženy nejvíce. Postižení je multisystémové, vynímečně je postižen jeden orgán (LHON). popisovány od r. 1988 čtyři komplexy dýchacích enzymů a komplexu F1F0 ATP syntázy - oxidativní fosforylace,
 jsou postiženy nejvíce. Postižení je multisystémové, vynímečně je postižen jeden orgán (LHON). popisovány od r čtyři komplexy dýchacích enzymů a komplexu F1F0 ATP syntázy - oxidativní fosforylace,")
38
s progredujícím průběhem a postihující zdánlivě nesouvisející orgány.
Defekt v mitochondriálním respiračním řetězci by se měl zvažovat u pacientů, kteří mají jakoukoli nevysvětlenou kombinaci neuromuskulárních a/nebo jiných příznaků, s progredujícím průběhem a postihující zdánlivě nesouvisející orgány. Munnich et al, OMMBD, chapter 99

39
Mitochondriální, maternální dědičnost

40
Mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes (MELAS) - demence, ztráta sluchu, malý vzrůst, mrtvice v mladém věku Leber hereditary optic neuropathy – LHON - degenerace ganglionů sítnice, atrofie očního nervu, ztráta vidění Dědičné metabolické poruchy mitochondrií: Leighův syndrom • Chronická progresivní externí oftalmoplegie • Syndrom Kearns-Sayre • Leberova hereditární optická neuropatie • Maternálně dědičný diabetes a hluchota • Deficit pyruvát dehydrogenázy • Deficit pyruvát karboxylázy • Deficit fosfoenolkarboxykinázy MELAS hypotonie, hypertrophic cardiomyopathy (HCMP), mléčná acidóza, hypospadie, hyperammonaemia a 3-methylglutaconic aciduria LHON degenerace ganglionů sítnice buňky (RGCs) a jejich neuritech
, mléčná acidóza, hypospadie, hyperammonaemia a 3-methylglutaconic aciduria. LHON degenerace ganglionů sítnice buňky (RGCs) a jejich neuritech.")
41
Lokalizace nejčastějších mutací MELAS, MERRF, LHON, NARP syndromu
Mitochondriální encefalopatie, laktátová acidóza a syndrom cerebrovaskulárních nehod (MELAS) Je hlavně asociováno s mutací (A3243G) mtDNA v genu pro tRNA Leu (UUR). Genetický kód mitochondrie a eukarytoního jádra je totožný s vyjímkou čtyř kodónů. 4 triplety mají jiný význam proti jadernému genomu, rozdíly spočívají v iniciaci a terminaci UGA codon is not a termination codon, but codes for tryptophan, on the other hand codons AGA and AGG do not code for leucine, but terminate translation and the codon AUA codes for methionine instead for leucine. Lokalizace nejčastějších mutací MELAS, MERRF, LHON, NARP syndromu
 Je hlavně asociováno s mutací (A3243G) mtDNA v genu pro tRNA Leu (UUR). Genetický kód mitochondrie a eukarytoního jádra je totožný s vyjímkou čtyř kodónů. 4 triplety mají jiný význam proti jadernému genomu, rozdíly spočívají v iniciaci a terminaci. UGA codon is not a termination codon, but codes for tryptophan, on the other hand codons AGA and AGG do not code for leucine, but terminate translation and the codon AUA codes for methionine instead for leucine. Lokalizace nejčastějších mutací MELAS, MERRF, LHON, NARP syndromu.")
42
Mutace v mtDNA Variabilní expresivita typická pro mitochondriální choroby – při dělení je distribuce do dceřinných buněk náhodná, proto rozdělení mutované a normální DNA je různé Homoplazmie = buň mají jen mutantní mtDNA nebo jen normální mtDNA Heteroplazmie v buňce je směs normální a mutantní mtDNA Prenatal genetic testing and interpretation of test results for mtDNA disorders are difficult

43
Mutace v mtDNA Mutace cca 10x častěji než v jaderné DNA, protože chybí histony a některé reparační mechanizmy. Přibližně 10% všech mitochondriálních proteinů jsou kódovány v jádře. V případě mutací v těchto genech – AR dědičnost Některé mitochondriální proteiny jsou agregáty původem z jaderných a mitochondriálních genů čerstvé mutace vznikají pravidelně, v dané sekvenci asi 1krát za 1500–3000 let

44
Děkuji za pozornost Použitá literatura: Thompson and Thompson: Klinická genetika, 6. Vydání, 2004 Adkinson J R, Brown M.D.: Elsevier‘s Integrated Genetics, 2007

Podobné prezentace












>")






 vylučované kůrou nadledvinek (aldosteron, kortisol); 2) vylučované pohlavními žlázami (progesteron, testosteron, estradiol)>")
