Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Metabolismus aminokyselin I
Jana Novotná 2. LF UK, Ústav lékařské chemie a klinické biochemie

2
Osnova přednášky Přehled metabolismu aminokyselin
Trávení a absorpce, přenos AK do buněk Obecné reakce aminokyselin (transaminace, deaminace, dekarboxylace) Syntéza neesenciálních aminokyselin Katabolismus aminokyselin
 Syntéza neesenciálních aminokyselin. Katabolismus aminokyselin.")
3
Metabolismus aminokyselin
Tělesné proteiny 300 – 400 g/den Proteosyntéza Proteolýza Nebílkovinné deriváty: porfyriny puriny pyrimidiny neurotransmitery hormony složené lipidy aminocukry Trávení Proteiny z potravy zásoba volných aminokyselin Transaminace Přeměna ulíkové kostry Glykolýza krebsův cyclus Močovina NH3 Sacharidy lipidy CO2 voda Ketolátky Acetyl CoA

4
Trávicí enzymy štěpící proteiny
Trávicí trakt Endopeptidasy - žaludeční - pepsin Pankreatické - trypsin, chymotrypsin, elastasa Exopeptidasy (pankreas) - aminopeptidasy, karboxypeptidasy, dipeptidasy Hydrolýza polypetidy oligopetidy aminokyseliny střevní sliznice transport k cílovým tkáním. Pepsin (pH 1.5 – 2.5) – peptidová vazba mezi hydrofóbními AK a aromatickými AK (Tyr, Phe, Trp) Trypsin (pH 7.5 – 8.5) – peptidová vazba za Lys a Arg Chymotrypsin (pH 7.5 – 8.5) – peptidová vazba za aromatickými AK (Trp, Phe,Tyr), v menší míře za Met, Leu Pankreatická elastasa (pH 7.5 – 8.5) – peptidová vazba za Ala, Gly a Ser Odbourávání aminokyselin intracelulárně Prvním krok - odstranění a-aminoskupiny většinou jako NH3 vyloučen přímo nebo přes další sloučeniny.
 - aminopeptidasy, karboxypeptidasy, dipeptidasy. Hydrolýza polypetidy oligopetidy aminokyseliny střevní sliznice transport k cílovým tkáním. Pepsin (pH 1.5 – 2.5) – peptidová vazba mezi hydrofóbními AK a aromatickými AK (Tyr, Phe, Trp) Trypsin (pH 7.5 – 8.5) – peptidová vazba za Lys a Arg. Chymotrypsin (pH 7.5 – 8.5) – peptidová vazba za aromatickými AK (Trp, Phe,Tyr), v menší míře za Met, Leu. Pankreatická elastasa (pH 7.5 – 8.5) – peptidová vazba za Ala, Gly a Ser. Odbourávání aminokyselin intracelulárně. Prvním krok - odstranění a-aminoskupiny většinou jako NH3 vyloučen přímo nebo přes další sloučeniny.")
5
Vstřebávání aminokyselin ve střevě
Kotransport s Na+ - semispecifické Na+ dependentní proteiny AK se dostává na serózní straně usnadněným transportem po koncentračním spádu Na+ (sodíkový gradient). Transportní systémy pro neutrální aminokyseliny, prolin a hydroxyprolin, kyselé, bazické aminokyseliny a cystin.
. Transportní systémy pro neutrální aminokyseliny, prolin a hydroxyprolin, kyselé, bazické aminokyseliny a cystin.")
6
Klinická korelace Geneticky podmíněný porušený transport aminokyselin do buněk kartáčového lemu tenkého střeva a ledvinových tubulů, není reabsorpce v proximálním tubulu aminokyseliny do moče Cystinurie – přenašeč pro cystin a bazické aminokyseliny, lysin, arginin, ornithin do buněk: ledvinové kameny Hartnupova nemoc - vrozená izolovaná porucha transportu neutrálních aminokyselin střevní sliznicí a renálními tubuly podmíněná defektem specifického transportního genu pro tyto aminokyseliny na 2. chromozomu obvykle nevyvolává žádné klinické příznaky, jen malá část pacientů kolem 10 roku fotosenzitivní, ekzémy. Moč novorozenců je rutinně vyšetřována

7
Intracelulární metabolický obrat proteinů a doplňování zásoby volných aminokyselin
Poločas metabol. obratu proteinů: krátkodobé p. (regulační, špatně složené) – poločas minuty dlouhodobé p. (většina buněčných proteinů) – poločas dny, týdny strukturální p. (kolagen) – metabolicky stabilní Odbourání proteinů: systém ATP-dependentního ubikvitinového proteazomu (cytosol) endogenní proteiny systém na ATP nezávislých lysozomálních enzymů (kyselé hydrolasy atd.) extracelulární, povrchově membránové p.
 – poločas minuty. dlouhodobé p. (většina buněčných proteinů) – poločas dny, týdny. strukturální p. (kolagen) – metabolicky stabilní. Odbourání proteinů: systém ATP-dependentního ubikvitinového proteazomu (cytosol) endogenní proteiny. systém na ATP nezávislých lysozomálních enzymů (kyselé hydrolasy atd.) extracelulární, povrchově membránové p.")
8
Proteázy podílející se na metabolismu/odbourávání proteinů
Klasifikace Mechanismus Úloha Katepsiny Cysteinové proteasy (cystein v aktivním místě) Lysosomální enzymy Kaspasy Cysteinové proteasy štěpící za aspartátem Apoptosa; aktivované z prokaspas Matrixové metaloproteinasy Zinek jako kofaktor Degradace extracelulární matrix, regulace inhibitory TIMPs (tissue inhibitors of matrix metalloproteiinases) Proteazom Velký komplex – degradace ubiquitinem značené proteiny Metabolický obrat proteinů Serinové proteasy Serin v aktivním místě společně s histidinem a aspartátem Mnoho enzymů odbourávání proteinů; koagulace, aktivované ze zymogenů Kalpainy Cysteinové proteasy, vápník jako kofaktor Řada úloh v buněčné metabolismu
 Lysosomální enzymy. Kaspasy. Cysteinové proteasy štěpící za aspartátem. Apoptosa; aktivované z prokaspas. Matrixové metaloproteinasy. Zinek jako kofaktor. Degradace extracelulární matrix, regulace inhibitory TIMPs (tissue inhibitors of matrix metalloproteiinases) Proteazom. Velký komplex – degradace ubiquitinem značené proteiny. Metabolický obrat proteinů. Serinové proteasy. Serin v aktivním místě společně s histidinem a aspartátem. Mnoho enzymů odbourávání proteinů; koagulace, aktivované ze zymogenů. Kalpainy. Cysteinové proteasy, vápník jako kofaktor. Řada úloh v buněčné metabolismu.")
9
Ubikvitin-proteazom

10
Glutathion a přenos aminokyselin (gama-glutamylový cyklus)
g-glutamyltranserasa (g-glutamyl transpeptidasa GGT) Výskyt GGT - membrány buněk s vysokou sekreční nebo absorpční kapacitou. Játra – mikrozomální frakce hepatocytů a membrány buněk výstelky žlučových cest Proximální tubuly ledvin, enterocyty, pankreas GGT má diagnostický význam u hepatobiliárních poruch. Převzato z
 Výskyt GGT - membrány buněk s vysokou sekreční nebo absorpční kapacitou. Játra – mikrozomální frakce hepatocytů a membrány buněk výstelky žlučových cest. Proximální tubuly ledvin, enterocyty, pankreas. GGT má diagnostický význam u hepatobiliárních poruch. Převzato z")
11
Obecné reakce aminokyselin
Transaminace Deaminace Oxidativní dekarboxylace

13
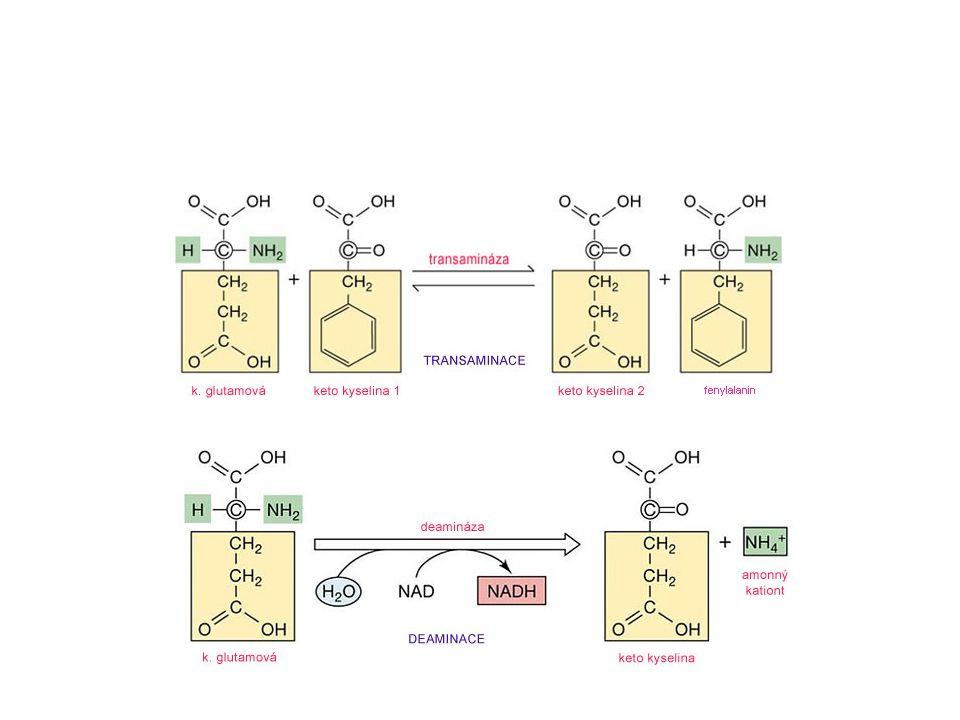
Transaminace Aminokyselina ztrácí aminoskupinu - enzymová katalýza, transaminace (transaminasa = aminotransferasa) Transaminační reakce probíhají u všech AK s výjimkou lysinu, threoninu a prolinu Většina transaminas vyžaduje a-ketoglutarát jako příjemce aminoskupiny Větvené aminokyseliny mají společnou transaminasu

14
Transaminasy - specifické enzymy vždy pro jeden pár aminokyseliny s její odpovídající a-ketokyselinou (oxokyselinou). Prostetická skupina – pyridoxalfosfát (PLP)
")
15
Interní Schiffova báze
PLP - pevně vázán na enzym (transaminasu) Schiffovou bází na e-aminoskupinu lysinu. N H + CH3 O- C H2C 2-O3PO Enzym Lysin Interní Schiffova báze
 Schiffovou bází na e-aminoskupinu lysinu. N. H. + CH3. O- C. H2C. 2-O3PO. Enzym. Lysin. Interní Schiffova báze.")
16
Sled reakcí při transaminaci

17
Transaminasy důležité v klinické praxi
AST - aspartátaminotransferasa, starší název SGOT (serum glutamát- oxaloacetáttransaminasa). V játrech, v kosterním a srdečním svalu, méně i v jiných parenchymatózních orgánech, v erytrocytech Nachází se jak v cytoplasmě, tak v mitochondriích Zvýšení akutní infarkt myokardu onemocnění kosterních svalů akutní hepatitida a jiné hepatocelulární léze
. V játrech, v kosterním a srdečním svalu, méně i v jiných parenchymatózních orgánech, v erytrocytech. Nachází se jak v cytoplasmě, tak v mitochondriích. Zvýšení. akutní infarkt myokardu. onemocnění kosterních svalů. akutní hepatitida a jiné hepatocelulární léze.")
18
Transaminázy důležité v klinické praxi
ALT- alaninaminotransferasa starší název SGPT (serum glutamát- pyruváttransferasa). Čistě cytoplasamtický enzym, hlavně v játrech Indikátor poškození hepatocytu (jaterních léze) Zvýšení virová hepatitida mononukleóza
. Čistě cytoplasamtický enzym, hlavně v játrech. Indikátor poškození hepatocytu (jaterních léze) Zvýšení. virová hepatitida. mononukleóza.")
19
Deaminace aminokyselin
aminokyselina FMN H2O + a-ketokyselina FMNH2 NH3 oxidasa L-aminokyselin A. Oxidativní deaminace H2O2 O2 katalasa B. Neoxidativní deaminace serin pyruvát threonin a-ketobutyrát NH3 + H2O serin-threonin dehydratasa Oxidáza L-aminokyselin tvoří NH3 a a-ketokyselinu přímo, FMN jako kofaktor.

20
Dekarboxylace Katalyzována dekarboxylasami
Kofaktor pyridoxalfosfát R-CHNH2-COOH R-CH2NH2 + CO2 Probíhá v malých množstvích Primární aminy Biologicky aktivní aminy Hormony (neurotransmitery, koenzymy)
")
21
Přehled biogenních aminů

22
Syntéza neesenciálních aminokyselin

23
Přehled syntézy neesenciálních aminokyselin
10 AK – z glukózy přes intermediáty glykolýzy nebo citrátového cyklu Phe Tyr Cys Ser + síra z Met

24
Aminokyseliny odvozené z intermediátů glykolýzy

25
Serin Serin: – inhibice 3-fosfoglycerát- dehydrogenázy
- ihibice fosfoserinfosfatázy

26
Vzájemná přeměna serinu a glycinu
Převzato z

27
Odbourání glycinu Glycinsyntasa (H4folate) Převzato z
 Převzato z")
28
Tetrahydrofolát jako nosič jednoho uhlíku
Kyselina listová dihydrofolátreduktasa – NADPH (2x) Tetrahydrofolát Serin glycin – vznik N5,N10-methylen THF Glycin CO2 - vznik N5,N10-methylen THF Homocystein methionin – donor N5-methyl THF Histidin odbourání – vznik N5-formiminoTHF; N5,N10-metnhenyl a N10-formyl THF Tryprofan odbourání – vznik N10-formyl THF
 Tetrahydrofolát. Serin glycin – vznik N5,N10-methylen THF. Glycin CO2 - vznik N5,N10-methylen THF. Homocystein methionin – donor N5-methyl THF. Histidin odbourání – vznik N5-formiminoTHF; N5,N10-metnhenyl a N10-formyl THF. Tryprofan odbourání – vznik N10-formyl THF.")
29
Souhrn metabolismu glycinu

30
Aminokyseliny vztahující se k oxalacetátu
Aspartát a asparagin

31
*nefunkční enzym vede ke vzniku homocystinurie
Biosyntéza cysteinu Regenerace Met za přítomnosti N5-methyl- tetrahydrofolátu (vitaminy: folát + B12) SAM se přes SAH mění na homocystein. Homocystein kondenzuje se serinem na cystathion. Cystathionasa rozštěpí cystathion na cystein a a-keto-glutarát. * Celá rerakce se nazývá transsulfurace *nefunkční enzym vede ke vzniku homocystinurie Převzato z
 SAM se přes SAH mění na homocystein. Homocystein kondenzuje se serinem na cystathion. Cystathionasa rozštěpí cystathion na cystein a a-keto-glutarát. * Celá rerakce se nazývá transsulfurace. *nefunkční enzym vede ke vzniku homocystinurie. Převzato z")
32
Homocystinurie Klinická poznámka
Vrozená porucha metabolismu Met, geneticky podmíněná defektem enzymu cystathionin-β-synthasy. V moči je vysoká koncentrace homocysteinu a methioninu. Deformity kostí, poruchy zraku způsobené atypickým uložením čočky, předčasná ateroskleróza, hluboká žilní tromboembolie, postižení CNS. Neléčený stav vede k opožděnému mentálnímu vývoji. Vysoká chemická reaktivita homocysteinu a působení vzniku volných radikálů narušují jiné enzymy a mitochondrie buněk.

33
Syntéza a degradace prolinu

34
Vzájemná přeměna mezi glutamátem, glutaminem a a-ketoglutarátem
NH3 NH3 a-ketoglutarát glutamát glutamin NH3 NH3 A. Glutamátdehydrogenasa glutamát + + NAD+ H2O a-ketoglutarát + NH3 + NADH z transaminačních reakcí přímo do močovin. cyklu B. Glutaminsyntetasa (játra) ATP ADP + glutamát NH3 Glutamin (puriny, pyrimidiny, regulace pH) C. Glutaminasa (ledviny) + + glutamin H2O glutamát NH3
 ATP. ADP. + glutamát. NH3. Glutamin (puriny, pyrimidiny, regulace pH) C. Glutaminasa (ledviny) + + glutamin. H2O. glutamát. NH3.")
35
Degradace aminokyselin

36
Dvacet aminokyselin se odbourává na sedm produktů, které jsou součástí citrátového cyklu
pyruvát acetyl CoA oxalacetát fumarát sukcinyl CoA acetoacetyl CoA a-ketoglutarát citrát PEP aspartát asparagin tyrosin fenylalanin isoleucin methionin threonin valin alanin, glycin cystein, serin tryptofan leucin* leucin*,lysin* tyrosin, tryptofan glukóza lipidy glutamin glutamát histidin prolin arginin

37
Glukogenní aminokyseliny
a-ketoglutarát, pyruvát, oxaloacetát, fumarát nebo sukcinyl CoA Aspartát Asparagin Arginin Phenylalanin Tyrosin Isoleucin Methionin Valin Glutamin Glutamát Prolin Histidin Alanin Serin Cystein Glycin Threonin Tryptofan

38
Ketogenní aminokyseliny
Acetyl CoA nebo acetoacetát Lysin Leucin

39
Ketogenní a glukogenní aminokyseliny
a-ketoglutarát, pyruvát, oxaloacetát, fumarát nebo sukcinyl CoA a také acetyl CoA nebo acetoacetát Isoleucin Threonin Tryptofan Fenylalanin Tyrosin

40
Aminokyseliny tvořící sukcinyl CoA

41
Aminokyseliny tvořící acetyl CoA a acetoacetát

42
Aminokyseliny skupiny glutamátu

43
Odbourání histidinu

44
Metabolismus methioninu
Tvorba aktivovaného methioninu = S-adenosylmethionin (SAM) SAM slouží jako prekurzor pro řadu metylačních reakcí, např. konverze noradrenlinu na adrenalin. Po ztrátě CH3 vzniká S-adenosylhomocystein (SAH). Methionin homoserin propionyl-CoA methylmalonyl-CoA sukcinyl-CoA
 SAM slouží jako prekurzor pro řadu metylačních reakcí, např. konverze noradrenlinu na adrenalin. Po ztrátě CH3 vzniká S-adenosylhomocystein (SAH). Methionin homoserin propionyl-CoA methylmalonyl-CoA sukcinyl-CoA.")
45
Odbourávání větvených aminokyselin

46
Aminoacidémie větvených aminokyselin, leucinóza
Klinická poznámka Aminoacidémie větvených aminokyselin, leucinóza (choroba javorového sirupu) Vrozená genetická porucha metabolismu větvených aminokyselin, geneticky podmíněná defektem enzymu dehydrogenasa větvených a-ketokyselin. Větvené aminokyseliny a jejich a-ketokyseliny se dostávají ve vysokých koncentracích do moči. Mechanismus toxicity není znám. Neléčený stav vede k abnormálnímu vývoji mozku a mentální retardaci.
 Vrozená genetická porucha metabolismu větvených aminokyselin, geneticky podmíněná defektem enzymu dehydrogenasa větvených a-ketokyselin. Větvené aminokyseliny a jejich a-ketokyseliny se dostávají ve vysokých koncentracích do moči. Mechanismus toxicity není znám. Neléčený stav vede k abnormálnímu vývoji mozku a mentální retardaci.")
47
Bioyntéza tyrosinu z fenylalaninu
Tetrabiopterin redukuje fenylalaninhydroxylasu a sám je zpět redukován NADH-dependentní dihydropteridinreduktasou. Chybějící nebo defektní fenylalaninhydroxylasa způsobuje hyperfenylalaninemie (koncentrace Phe > 120 mM). Převzato z
. Převzato z")
48
Tetrahydrobiopterin jako kofaktor hydroxylas
Dihydrobiopterin

49
Klinická poznámka Fenylketonurie
Vrozená porucha metabolismu Phe, geneticky podmíněná defektem enzymu fenylalaninhydroxyláza (chromosom 12) Nahromaděný Phe (1000 mM v plasmě) se stává hlavním donorem aminoskupiny a odčerpává v nervové tkáni a-ketoglutarát. V nervové tkáni chybí a-ketoglutarát pro Krebsův cyklus, snižuje se aerobní metabolismus. Neléčený stav vede k mentální retardaci.
 Nahromaděný Phe (1000 mM v plasmě) se stává hlavním donorem aminoskupiny a odčerpává v nervové tkáni a-ketoglutarát. V nervové tkáni chybí a-ketoglutarát pro Krebsův cyklus, snižuje se aerobní metabolismus. Neléčený stav vede k mentální retardaci.")
50
Tryptofan Otevření pyrrolového kruhu (tryptofan-2,3-dioxygenasa).
Indolový kruh je ketogenní (acetoacetát). Odštěpení alaninu. Kynurenin – přeměna na několik produktů vylučovaných do moči (kyselina xanthurenová) Tryptofan je prekurzorem pro serotonin a melatonin.
. Odštěpení alaninu. Kynurenin – přeměna na několik produktů vylučovaných do moči (kyselina xanthurenová) Tryptofan je prekurzorem pro serotonin a melatonin.")
51
Selenocystein Nadávno zařazen mezi proteinogenní aminokyseliny jako 21 AK. Nachází se v aktivním místě různých enzymů, včetně antioxidačního enzymu glutathionperoxidasy a 5-deiodinas. Do proteinu se inkorporuje tRNA s UCA antikodonem. Záměna selenocysteinu za Cys vede ke značnému snížení enzymové aktivity (nedostatek Se v potravě).
.")
52
Schémata použitá v prezentaci:
Marks´ Basic Medical Biochemistry A Clinical Approach. Four edition M. Lieberman, A.D. Marks ed., 2013. Essentials of Medical Biochemistry With Clinical Cases. First edition. N.V. Bhagavan, Chung-Eun Ha ed., 2011. Zdroje z internetu jsou uvedené u jednotlivých schémat.

Podobné prezentace