Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Dědičné periferní neuropatie – Charcot-Marie-Tooth (CMT/HMSN) co to je
Dědičné periferní neuropatie – Charcot-Marie-Tooth (CMT/HMSN) co to je?, jak se to projevuje? , jak se to dědí?, jak se to diagnostikuje?, jak se to lečí? P. Seeman Klinika dětské neurologie, DNA laboratoř, UK 2. LF a FN Motol Praha
 co to je , jak se to projevuje , jak se to dědí , jak se to diagnostikuje , jak se to lečí P. Seeman Klinika dětské neurologie, DNA laboratoř, UK 2. LF a FN Motol Praha.")
2
Co je C-M-T ? Charcot a Marie – Francouzi, Tooth - Angličan – lékaři, kteří ve druhé polovině předminulého století (1886) poprvé popsali tuto chorobu – dědičnou neuropatii – tzv. peroneální svalovou atrofii Termín CMT (HMSN) zastřešuje různé typy dědičných neuropatií – odlišnosti jsou ve způsobu dědičnosti, věku při objevení se choroby, elektrofyziologických nálezech, biopsii nervu, molekulárně genetických nálezech atd.
 poprvé popsali tuto chorobu – dědičnou neuropatii – tzv. peroneální svalovou atrofii. Termín CMT (HMSN) zastřešuje různé typy dědičných neuropatií – odlišnosti jsou ve způsobu dědičnosti, věku při objevení se choroby, elektrofyziologických nálezech, biopsii nervu, molekulárně genetických nálezech atd.")
3
Dědičná neuropatie CMT
Nejčastější dědičné nervosvalové onemocnění Projevuje se oslabením distálních – (periferních) svalů končetin v důsledku postižení periferních nervů Oslabení svalů se obvykle pomalu šíří na proximálnější svaly (blíže k trupu) Pacienti většinou trpí zhoršenou chůzí a poruchou citlivosti končetin
 svalů končetin v důsledku postižení periferních nervů. Oslabení svalů se obvykle pomalu šíří na proximálnější svaly (blíže k trupu) Pacienti většinou trpí zhoršenou chůzí a poruchou citlivosti končetin.")
4
Klinický obraz C-M-T Incidence 1 : : v ČR asi – lidí manifestace většinou dekáda – později vzácně vždy distální svalová slabost a atrofie na DK často deformity nohou a skoliosa většinou poruchy čití na DK vyhaslé či snížené reflexy na DK velká variabilita kliniky i v jedné rodině

5
Jak se CMT projevuje Obvyklé postižení svalů přední strany bérce – peroneálních svalů – přepadávání špičky nohy - zakopávání vázne či není možná chůze po patách

6
Jak se CMT projevuje. Slábnutím distálních svalů, atrofie –deformity, poruchy chůze, poruchy citlivosti a další

7
-atrofie lýtek, -varosní deformity nohou, -atrofie malých svalů rukou

8
Minimální postižení u 16 leté dívky s CMT1A duplikací
-oslabení a omezení extenze nohy -nášlap na celé chodidlo -neschopnost chůze po patách -normální ruce

9
Dědičné neuropatie - CMT
HMSN - Hereditary Motor and Sensory Neuropathies HMN - Hereditary Motor Neuropathies ( distal SMA) HSN/HSAN - Hereditary Sensory Neuropathies P.J. Dyck
 HSN/HSAN - Hereditary Sensory Neuropathies. P.J. Dyck")
10
Hlavní typy CMT (patofyziologicky)
Při neuropatii může být primárně postižen buď : obal – myelin – tzv. demyelinizační neuropatie (hypertrofická) CMT typ 1- většina nebo vlákno samo – axon – axonální neuropatie – CMT typ vzácněji obvykle ale podíl obou, protože jsou na sobě závislé axonální neuropatie cibulovité formace tomakulum
 CMT typ 1- většina nebo vlákno samo – axon – axonální neuropatie – CMT typ 2 vzácněji obvykle ale podíl obou, protože jsou na sobě závislé. axonální neuropatie. cibulovité formace. tomakulum.")
11
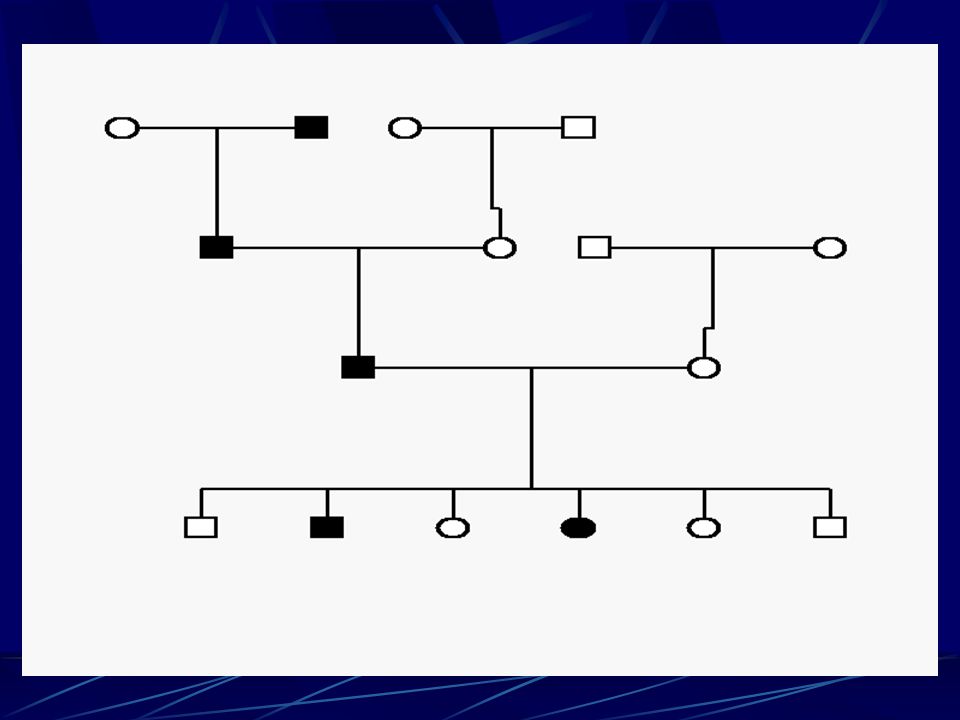
Jak se CMT dědí ? - typy dědičnosti
Dominantní způsob ( bez vazby na pohlaví) – postižený má pravděpodobnost 50% že předá tuto vlohu na svým dětem – je jedno jaké pohlaví – většina CMT se dědí tímto způsobem Dominantní způsob vázaný na pohlavní chromozom X – postižený muž nemůže předat tuto vlohu svému synovi ( nepředává mu svůj X chromozom, ale předá vlohu všem svým dcerám (100%) – ženy jsou však většinou postiženy méně a později – v průměru Recesívní (bez vazby na pohlaví) – relativně vzácný, postižený má jen minimálně zvýšené riziko, že budou stejně postiženy i jeho děti (partner by musel být buď stejně postižen s poruchou stejného genu nebo aspoň být nosič jedné poruchy ve stejném genu – minimální pravděpodobnost) Sporadický – nikdo v rodině nikde nemá podobné postižení – těžké dělat závěry – může být recesívní, ale a častěji jde o novou mutaci, která se pak dědí dominantně – tedy pro děti postiženého je 50 % riziko
 – postižený má pravděpodobnost 50% že předá tuto vlohu na svým dětem – je jedno jaké pohlaví – většina CMT se dědí tímto způsobem. Dominantní způsob vázaný na pohlavní chromozom X – postižený muž nemůže předat tuto vlohu svému synovi ( nepředává mu svůj X chromozom, ale předá vlohu všem svým dcerám (100%) – ženy jsou však většinou postiženy méně a později – v průměru. Recesívní (bez vazby na pohlaví) – relativně vzácný, postižený má jen minimálně zvýšené riziko, že budou stejně postiženy i jeho děti (partner by musel být buď stejně postižen s poruchou stejného genu nebo aspoň být nosič jedné poruchy ve stejném genu – minimální pravděpodobnost) Sporadický – nikdo v rodině nikde nemá podobné postižení – těžké dělat závěry – může být recesívní, ale a častěji jde o novou mutaci, která se pak dědí dominantně – tedy pro děti postiženého je 50 % riziko.")
12
Jak se CMT dědí ? - typy dědičnosti
Dominantní způsob ( bez vazby na pohlaví) – postižený má pravděpodobnost 50% že předá tuto vlohu na svým dětem – je jedno jaké pohlaví – většina CMT se dědí tímto způsobem
 – postižený má pravděpodobnost 50% že předá tuto vlohu na svým dětem – je jedno jaké pohlaví – většina CMT se dědí tímto způsobem.")
14
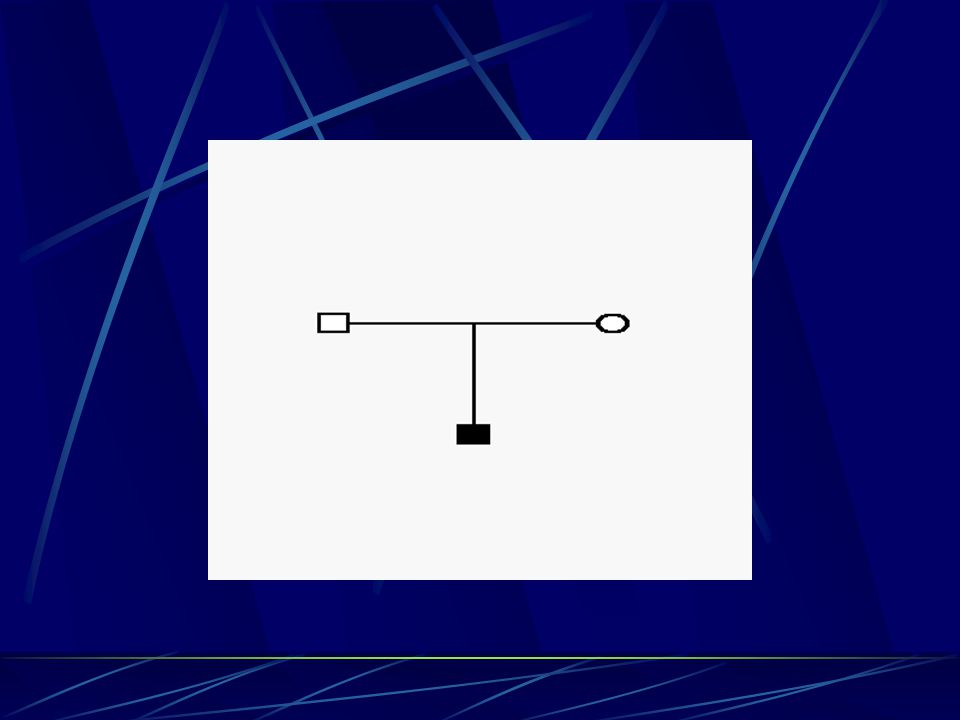
Jak se CMT dědí ? - typy dědičnosti
Dominantní způsob vázaný na pohlavní chromozom X – postižený muž nemůže předat tuto vlohu svému synovi ( nepředává mu svůj X chromozom, ale předá vlohu všem svým dcerám (100%) – ženy jsou však většinou postiženy méně a později – v průměru
 – ženy jsou však většinou postiženy méně a později – v průměru.")
16
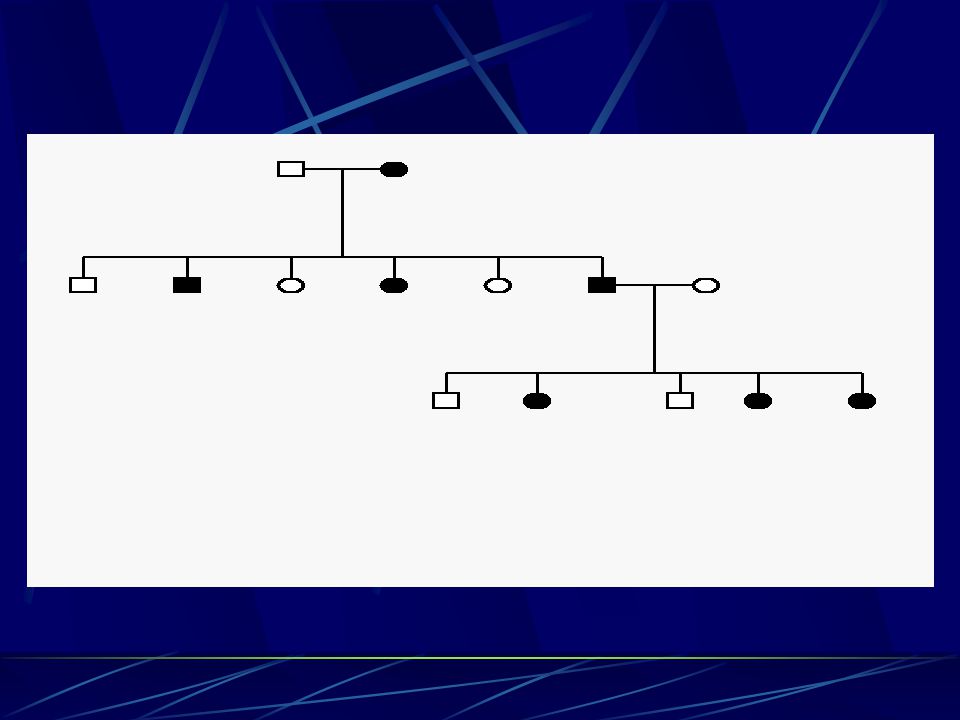
Jak se CMT dědí ? - typy dědičnosti
Recesívní (bez vazby na pohlaví) – relativně vzácný, postižený má jen minimálně zvýšené riziko, že budou stejně postiženy i jeho děti (partner by musel být buď stejně postižen s poruchou stejného genu nebo aspoň být nosič jedné poruchy ve stejném genu – minimální pravděpodobnost)
 – relativně vzácný, postižený má jen minimálně zvýšené riziko, že budou stejně postiženy i jeho děti (partner by musel být buď stejně postižen s poruchou stejného genu nebo aspoň být nosič jedné poruchy ve stejném genu – minimální pravděpodobnost)")
18
Jak se CMT dědí ? - typy dědičnosti
Sporadický – nikdo v rodině nikde nemá podobné postižení – těžké dělat závěry – může být recesívní, ale a častěji jde o novou mutaci, která se pak dědí dominantně – tedy pro děti postiženého je 50 % riziko – velmi častý a složitý případ

20
Klasické fenotypy HMSN
netěžší kongenitální hypomyelinace Dejerine-Sottas / HMSN III Klasický CMT fenotyp CMT1A a CMT1B HNPP / 1,5 Mb delece nejlehčí

21
Jak se neuropatie vyšetřují ?
Neurologické vyšetření: Celkové vyšetření – síla jednotlivých svalů, výpadky, reflexy, citlivost – čití výkadky – jaké, jak moc a kde Rodinná anamnéza – má ještě někdo v rodině podobné obtíže – tvar nohy atd ?? Elektrofyziologické vyšetření – EMG – měření jak rychle vedou nervy elektrické vzruchy, jak vypadají proudy ze svalů atd. – odlišení postižení nervů nebo svalů Stav centrální nervové soustavy – jak vedou dráhy – evokované potenciály Někdy i biopsie nervu – v posledních letech díky možnosti molekulárně genetického vyšetření se většinou nedělá, jen v nejasných a složitých případech Nenajde-li se jiná příčina a dle výsledků – úvaha a závěr na dědičnou neuropatii

22
Elektrodiagnostika CMT
Potvrzení neuropatie – odlišení od myopatie či míšního či mozkového postižení stanovení typu CMT /demyel. x axonopatie/ - typ 1 nebo 2 odhalení subklinických forem a jedinců v rodině – potvrzení podobného postižení v rodině – tím potvrzení dědičné příčiny neuropatie – velmi zásadní

23
Genetické vyšetření (3 kroky)
1. Genetická konzultace a vysvětlení smyslu a možností vyšetření, sestavení rodokmenu, určení možných typů dědičnosti, souhlas, odběry 2. Molekulárně genetické vyšetření 3. Genetická konzultace – vysvětlení a sdělení výsledků a konsekvencí – dovyšetření dalších v rodině atd

24
Nejčastější genetické změny na DNA (genotypy) u CMT
 u CMT")
25
MPZ - myelin protein zero Po – CMT1B, DSS nebo CMT2
Hlavní a nejčastěji změněné geny u CMT (po vyloučení CMT1A duplikace na chrom 17p) Cx 32 - connexin-32 – CMTX MPZ - myelin protein zero Po – CMT1B, DSS nebo CMT2 PMP 22 - peripheral myelin protein – CMT1A Je již známo 34 genů jejichž porucha může vést k jedné či i více formám CMT, celkem jich je možná , většinou jde ale o vzácné příčiny.
 Cx 32 - connexin-32 – CMTX. MPZ - myelin protein zero Po – CMT1B, DSS nebo CMT2. PMP 22 - peripheral myelin protein – CMT1A. Je již známo 34 genů jejichž porucha může vést k jedné či i více formám CMT, celkem jich je možná , většinou jde ale o vzácné příčiny.")
26
Častost a četnost jednotlivých mutací u HMSN I/ C-M-T 1
Duplikace, delece genu pro PMP 22, 1,5 Mb duplikace v oblasti 17p, 70 % všech HMSN I, mezi autos.domin. HMSN I dokonce 84% mutace v genu Cx 32, asi 10 % všech HMSN, mutace v genu P0 ( MPZ), asi 5% všech HMSN, bodové mutace v genu PMP 22 - vzácné , častostí těchto mutací je dán postup při jejich detekci: CMT1A duplikace - Cx32 - P0 - PMP22 - další
, asi 5% všech HMSN, bodové mutace v genu PMP 22 - vzácné , častostí těchto mutací je dán postup při jejich detekci: CMT1A duplikace - Cx32 - P0 - PMP22 - další.")
27
Genetická prevence V rámci genetického poradenství – vysvětlení rizik a možností přenosu V principu je možná i tzv. prenatální diagnostika – ale jen na vyložené přání rodiny – normálně vzhledem k nezkrácené délce života je eticky sporná Prenatální diagnostika – je-li mutace v rodině již známá a je-li přání rodiny – lze během těhotenství zjistit, zda plod zdědil onu vlohu nebo ne – vloha nemusí nic říkat o následné tíži a rozsahu postižení dosud jsme prováděli pouze 1x

28
Rozšíření možností přímého genového vyšetření dalších forem CMT v posledních 2 letech
Možnosti pro vyšetření axonálních forem CMT (MPZ , NEFL, GDAP1) Možnosti pro vyšetření recesívních forem CMT (PRX, GDAP1, MTMR2, LMNA) Možnosti vyšetření HMN (GARS, Dynactin I, HSP22) Možnosti vyšetření HSN (NTRK I, RAB 7, SPTLC1)
 Možnosti pro vyšetření recesívních forem CMT (PRX, GDAP1, MTMR2, LMNA) Možnosti vyšetření HMN (GARS, Dynactin I, HSP22) Možnosti vyšetření HSN (NTRK I, RAB 7, SPTLC1)")
29
Prognoza u CMT Závisí na typu a na genovém defektu – nejen ale na tom, průběh různý Začátek obvykle v dekádě Jsou však typy i s velmi časným začátkem a i od narození Naopak jiné typy začnou až ve vyšší dospělosti – mohou mít pak ale rychlejší a závažnější průběh Průběh však v průměru u většiny velmi pomalý, ale trvalý CMT normálně nezkracuje očekávanou délku života

30
Jak se CMT/HMSN dá léčit ?
Poruchu genu dosud možné vyléčit není – CMT tedy v současnosti vyléčit nelze Léčit a pomát však lze Rehabilitace, protetika, režimová opatření, prevence škodlivin, vitamíny, ortopedické korekce V budoucnu je velká naděje mnohem účinnější léčby – pokrok je velmi rychlý a stále rychlejší – faktory pro výživu a přežívání nervových vláken atd.

31
Rehabilitace a protetika
Rehabilitační zařízení – na různých místech v ČR – vyšetření a návrh opatření rehabilitačním lékařem se zkušeností s CMT – ve FN Motol Praha – Klinika rehabilitace – prim. MUDr. O. Horáček. Možnost hospitalizace s vyšetřením, zaučením cvičení, navržení a zhotovení vložek či pomůcek, konzultace ortopeda, Viz dále Velmi prospěšné a vhodné jsou lázeňské pobyty – nejméně 1x za rok

32
Ortopedie Operace – na měkkých tkáních – přesuny šlach
Operace na kostech Ve FN Motol – MUDr. Pavel Smetana, doc. MUDr. Václav Smetana

33
Nezbytnosti pro DNA vyšetření pro C-M-T
Pečlivé neurologické vyšetření včetně systematického testování síly a projevů svalové slabosti, charakteru chůze, jemné motoriky, reflexy, citlivost Osobní anamnéza - věk při počátku obtíží ( 1., 2., 3., 4. dekáda ??) Pečlivá a cílená rodinná anamnéza - výskyt podobných obtíží u rodičů ?? u sourozenců ?? u jiných příbuzných ??, přenos nemoci z otce na syna ?? nejlépe je vidět a vyšetřit příbuzné osobně !! EMG - potvrzení neuropatie, určení typu neuropatie (axonální x demyel.), distribuce neuropatie Bez těchto údajů nemá smysl a nebude možné provádět složitá DNA vyšetření
 Pečlivá a cílená rodinná anamnéza - výskyt podobných obtíží u rodičů u sourozenců u jiných příbuzných , přenos nemoci z otce na syna nejlépe je vidět a vyšetřit příbuzné osobně !! EMG - potvrzení neuropatie, určení typu neuropatie (axonální x demyel.), distribuce neuropatie. Bez těchto údajů nemá smysl a nebude možné provádět složitá DNA vyšetření.")
34
Evropská i celosvětová spolupráce lékařů i vědců na problematice CMT
Evropské CMT consorcium – centrum v Antwerpách Severoamerické CMT consorcium Sdružení pacientů s CMT - Americká CMT Association

35
Děkuji za pozornost R. Mazanec O. Horáček A. Kobesová M. Bojar
P. Smetana A. Kobesová M. Bojar V. Beneš III. L. Baránková E. Mikešová J. Haberlová I. Sakmaryová

Podobné prezentace