Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Bruno Sopko

3
DMP=cca 1/10 vzácných nemocí Cca 700-800 nosologických jednotek (>1000 genů) Souhrnný výskyt nejméně 1:500 V obvodu každého praktického lékaře jsou nejméně 1-2 pacienti s DMP Cca 30% nemocí je léčitelných (100 nemocí dobře)
 Souhrnný výskyt nejméně 1:500 V obvodu každého praktického lékaře jsou nejméně 1-2 pacienti s DMP Cca 30% nemocí je léčitelných (100 nemocí dobře)")
5
< 1500 Da> 1500 Da Akutní toxicitaanone Chronická progrese±ano Lokalizacecytosol, ECTmembrány Strukturální dopadyneano Diagnostikakrev, močtkáně (moč) Původexogenníendogenní Léčba dieta, vitaminyúspěšnáneúčinná
 Původexogenníendogenní Léčba dieta, vitaminyúspěšnáneúčinná")
6
< 1500 Da ◦ plyny, anorganické ionty ◦ Aminokyseliny ◦ organické kyseliny ◦ Sacharidy ◦ Polyoly ◦ jednoduché lipidy ◦ puriny, pyrimidiny ◦ Vitaminy ◦ oligomery: peptidy do cca 5-10 AMK, oligosacharidy cytosol, stroma mitochondrií, krev, moč > 1500 Da ◦ Glykolipidy ◦ Sfingolipidy ◦ Plasmalogeny ◦ neutrální polysacharidy (glykogen) ◦ Mukopolysacharidy (další oligomery: proteiny, nukleové kyseliny...) obvykle asociované s membránami obvykle se nevyskytují ve větších množstvích v tělesných tekutinách ( x MS/MS technologie)
 ◦ Mukopolysacharidy (další oligomery: proteiny, nukleové kyseliny...) obvykle asociované s membránami obvykle se nevyskytují ve větších množstvích v tělesných tekutinách ( x MS/MS technologie)")
7
< 1500 Da obvykle závislé na exogenním přísunu manifestace (opakovanou) akutní toxicitou, často s encefalopathií/komatem častá hepatopathie časté odchylky v běžné biochemii- amoniak, ABR, ketolátky, glykemie, kys.močová... vznik symptomů závisí na specifické složce potravy, hladovění, katabolismu možný je i chronický průběh (při nízké toxicitě) obvykle relativně dobře léčitelné dietou a vitaminy > 1500 Da obvykle rozvoj bez závislosti na exogenním přísunu složek potravy typicky je průběh progresivní (± různě dlouhé bezpříznakové období) možná dysmorfie při porodu časté postižení nervového systému a svalstva organomegalie v důsledku střádání u lysosomálních nemocí obvykle neléčitelné dietou ani vitaminy
 obvykle relativně dobře léčitelné dietou a vitaminy > 1500 Da obvykle rozvoj bez závislosti na exogenním přísunu složek potravy typicky je průběh progresivní (± různě dlouhé bezpříznakové období) možná dysmorfie při porodu časté postižení nervového systému a svalstva organomegalie v důsledku střádání u lysosomálních nemocí obvykle neléčitelné dietou ani vitaminy.")
8
Novorozenecký screening je preventivní program, který slouží k vyhledávání novorozenců se zvýšeným rizikem některých onemocnění, u nichž lze při včasném odhalení a včasné léčbě předejít závažnému poškození zdraví dítěte. Vyšetření se provádí na základě příslušného metodického pokynu Ministerstva zdravotnictví ČR a je hrazeno ze zdravotního pojištění. Novorozenecký screening provádějí specializované laboratoře z tzv. suché kapky krve. Odběr několika kapek krve na speciální papírek se provádí ve věku 48 až 72 hodin po narození z patičky dítěte.

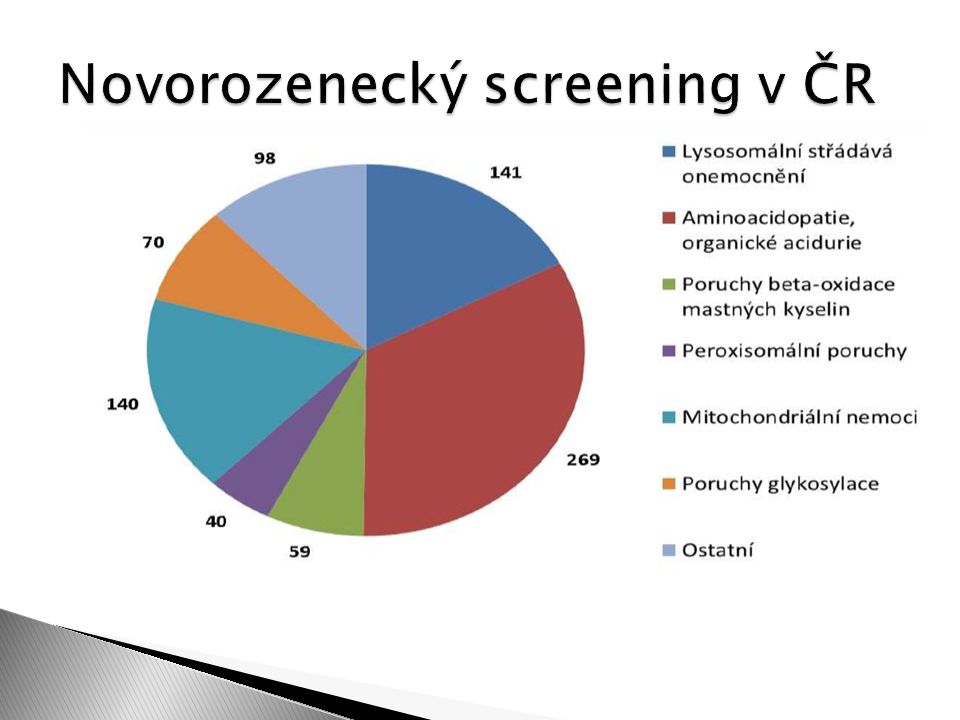
10
1. endokrinní onemocnění: ◦ kongenitální hypotyreóza (CH) ◦ kongenitální adrenální hyperplazie (CAH) 2. dědičné poruchy metabolismu: ◦ fenylketonurie (PKU) a hyperfenylalaninemie (HPA) ◦ nemoc javorového sirupu (MSUD) ◦ deficit acyl - CoA dehydrogenázy mastných kyselin se středně dlouhým řetězcem (MCAD) ◦ deficit 3 - hydroxyacyl - CoA dehydrogenázy mastných kyselin s dlouhým řetězcem (LCHAD) ◦ deficit acyl-CoA dehydrogenázy mastných kyselin s velmi dlouhým řetězcem (VLCAD) ◦ deficit karnitinpalmitoyltransferázy I (CPT I) ◦ deficit karnitinpalmitoyltransferázy II (CPT II) ◦ deficit karnitinacylkarnitintranslokázy (CACT) ◦ glutarová acidurie typ I (GA I) ◦ izovalerová acidurie (IVA) 3. jiná onemocnění: ◦ cystická fibróza (CF)
 ◦ kongenitální adrenální hyperplazie (CAH) 2. dědičné poruchy metabolismu: ◦ fenylketonurie (PKU) a hyperfenylalaninemie (HPA) ◦ nemoc javorového sirupu (MSUD) ◦ deficit acyl - CoA dehydrogenázy mastných kyselin se středně dlouhým řetězcem (MCAD) ◦ deficit 3 - hydroxyacyl - CoA dehydrogenázy mastných kyselin s dlouhým řetězcem (LCHAD) ◦ deficit acyl-CoA dehydrogenázy mastných kyselin s velmi dlouhým řetězcem (VLCAD) ◦ deficit karnitinpalmitoyltransferázy I (CPT I) ◦ deficit karnitinpalmitoyltransferázy II (CPT II) ◦ deficit karnitinacylkarnitintranslokázy (CACT) ◦ glutarová acidurie typ I (GA I) ◦ izovalerová acidurie (IVA) 3. jiná onemocnění: ◦ cystická fibróza (CF).")
11
OnemocněníEtnická skupinaIncidence (na 100000 narozených) Tay–Sachsova chorobaAškenázští Židé33 Gaucherova chorobaAškenázští Židé100 Hepatorenální tyrozinémieFrancouzsky mluvící oblast Kanady 54 Porfyria variegataBílí jihoafričané300 kongenitální adrenální hyperplazie (CAH) Jupici (eskymáci)200 fenylketonurie (PKU) a hyperfenylalaninemie (HPA) Turci Jemenitští Židé Aškenázští Židé Středoevropané 38,5 19 5 15 glutarová acidurie typ I (GA I)Odžibvejové Švédský původ > 50 3,3 nemoc javorového sirupu (MSUD)Mennonité568
 Tay–Sachsova chorobaAškenázští Židé33 Gaucherova chorobaAškenázští Židé100 Hepatorenální tyrozinémieFrancouzsky mluvící oblast Kanady 54 Porfyria variegataBílí jihoafričané300 kongenitální adrenální hyperplazie (CAH) Jupici (eskymáci)200 fenylketonurie (PKU) a hyperfenylalaninemie (HPA) Turci Jemenitští Židé Aškenázští Židé Středoevropané 38, glutarová acidurie typ I (GA I)Odžibvejové Švédský původ > 50 3,3 nemoc javorového sirupu (MSUD)Mennonité568")
12
Autozomálně recesivní Laboratorní diagnóza: 17-hydroxyprogesteron - zvýšen Léčba – podávání steroidů Dívky – chirurgická korekce

14
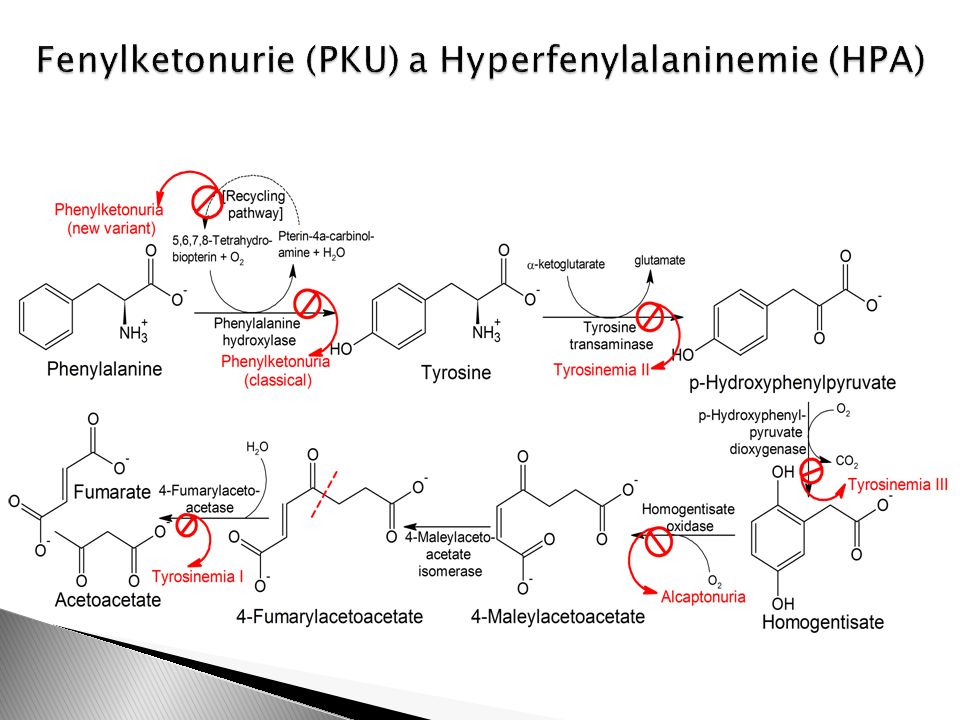
PKU HPA

15
Autozomálně recesivní Laboratorní diagnóza: zvýšený fenylalanin a poměr Phe/Tyr Léčba – alimentární – snížení příjmu Phe a dodání Tyr

16
Autozomálně recesivní Laboratorní diagnóza: Třístupňový model IRT/DNA/IRT: imunoreaktivního trypsinogenu (IRT) následná DNA analýza potní test Léčba: péče o dobrou průchodnost dýchacích cest: inhalace mukolytik + dechová fyzioterapie v několika(optimálně 3) krátkých blocích denně. péče o dobrý stav výživy: substituce pankreatických enzymů, vysokokalorická strava na 130 -150 %doporučených denních dávek pro běžnou populaci, suplementace vitamínů rozpustných v tucích, suplementace NaCl. kontrola infekce: antibiotika při každé akutní exacerbaci respirační infekce, širokospektrá, s protistafylokokovým účinkem; při záchytu Pseudomonas aeruginosa pak cílená ATB terapie proti tomuto patogenu, i když nejsou klinické příznaky. Dávkování vždy na horní hranici dávkovacího rozmezí, minimálně na 14 dnů.

17
http://www.novorozeneckyscreening.cz http://www.novorozeneckyscreening.cz Kožich, V. : Úvod do biochemické Genetiky Jean-Marie Saudubray, Georges van den Berghe, John H. Walter:Inborn Metabolic Diseases; Diagnosis and Treatment;Fifth Edition, Springer, 2012 William L. Nyhan, Bruce A. Barshop, Pinar T. Ozand, MD: Atlas of Metabolic Diseases; Second edition, Hodder Education,2005 Joe T. R. Clarke: A Clinical Guide to Inherited Metabolic Diseases;Second Edition, Cambridge University Press, 2004

Podobné prezentace





. Prognózování GPS a genetické poradenství Principem genetického prognózování je předpovědění vzniku určitého.>")



 Charakteristika látky rostlinného i živočišného původu deriváty vyšších mastných kyselin a alkoholu hydrofobní charakter ( odpuzují.>")










