Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Genetický podklad komplexních nemocí

2
Genom ve zdraví a nemoci
Genetická výbava jedince (souhrn všech genů=genom) je sice osudově zadána v okamžiku zplození, ale není pro další život konečná, protože v průběhu života se může měnit jak pod vlivem četných faktorů prostředí, tak pod vlivem dalších faktorů epigenetických.
 je sice osudově zadána v okamžiku zplození, ale není pro další život konečná, protože v průběhu života se může měnit jak pod vlivem četných faktorů prostředí, tak pod vlivem dalších faktorů epigenetických.")
3
Genomová stabilita vs. genomová variabilita
Stabilita = absence chyb Variabilita= adaptabilita genomu

4
Acetylace histonů jako podklad epigenetické modifikace chromatinu

5
Metylace DNA Metylace cytosinu je dědičná modifikace DNA v savčích buňkách a má determinující vliv na dlouhodobou represi genů a genomovou stabilitu. Genomické metylační „vzorce“, které zůstávají v dospělosti stabilní, se stávají hloubkově narušené v rakovinných buňkách většiny lidských tumorů. Mění se úroveň metylace v hyper i hypometylovaných oblastech genů. Ztráta metylace není patrně náhodná; patrně má mozaikový charakter a j e důsledkem přechodné demetylace.

6
Genové mutace Z hlediska patogeneze nemocí je důležité, zda se jedná o mutace v somatických buňkách, které vznikají v průběhu života, většinou jsou buněčně nebo tkáňově specifické a nepřenášejí se na potomstvo, nebo zda jde o tzv. zárodečné mutace, které vznikají v zárodečných buňkách (vajíčko nebo spermie), stávají se součástí vrozené genetické predispozice, jsou obsaženy ve všech buňkách a přenášejí se na potomstvo.
, stávají se součástí vrozené genetické predispozice, jsou obsaženy ve všech buňkách a přenášejí se na potomstvo.")
7
Genové mutace Mutací vzniklé alely jsou v populaci z různých důvodů vzácné (např. jsou výrazně patologické a tudíž jsou z populace odstraňovány selekcí, nebo vznikly nedávno a nestačily se v populaci rozšířit) a časté (polymorfismy).
 a časté (polymorfismy).")
8
Korelace fenotyp-genotyp u epidermolysis bullosa simplex (EBS)- příklad „monogenní „ nemoci, podmíněné vzácnými mutacemi Keratin 5 Keratin 14 Irvine AD, McLean WH The molecular genetics of the genodermatoses: progress to date and future directions. Br J Dermatol Jan;148(1):1-13. .
:")
9
Komplexní (multifaktoriální, multigenní) nemoci
musíme počítat s tím, že fakticky každá choroba má nějaké genetické pozadí, jehož podíl na manifestaci dané choroby je různý. Své genetické pozadí mají i tak relativně vzdálené proximální fenotypy, jako je např. kvalita života u nemocných s chronickým kardiovaskulárním onemocněním.

10
Komplexní (multifaktoriální, multigenní) nemoci
Za genetickou predispozici mnoha biologických procesů, evolučních adaptací a tedy také tzv. komplexních nemocí zřejmě odpovídají kombinace určitých genů a určitých faktorů zevního prostředí. Interakční efekty a vliv vnějších faktorů však nutně musíme očekávat i v případě mendelisticky děděných nemocí, což se koneckonců projevuje ve všeobecně známé lékařské zkušenosti se širším klinickým spektrem příznaků stejného onemocnění.

11
Komplexní (multifaktoriální, multigenní) nemoci
musíme počítat s tím, že fakticky každá choroba má nějaké genetické pozadí, jehož podíl na manifestaci dané choroby je různý. Své genetické pozadí mají i tak relativně vzdálené proximální fenotypy, jako je např. kvalita života u nemocných s chronickým kardiovaskulárním onemocněním.

12
Polymorfismy v DNA Jako polymorfismy v DNA se označují přirozeně se objevující změny v sekvenci DNA s více než jednou variantou-alelou, s populační frekvencí více než 1 %. Objevují se v průměru jednou na každých 1000 párů bází genomové DNA. Asi 90 % z nich jsou polymorfismy se záměnou jednoho nukleotidu (single nucleotide polymorphisms - SNP), jejichž podstatou je substituce jedné báze. Většina těchto polymorfismů leží v nekódujících (intronových) sekvencích, na jejichž funkční význam existují odlišné názory.
, jejichž podstatou je substituce jedné báze. Většina těchto polymorfismů leží v nekódujících (intronových) sekvencích, na jejichž funkční význam existují odlišné názory.")
13
Polymorfismy v DNA Kromě SNP se vyskytují také minisatelitní a mikrosatelitní polymorfismy, které vznikají v důsledku variace v tzv. tandemových repetitivních sekvencích. Minisatelitní polymorfismy jsou obvykle dlouhé 0,1-20 kilobází, zatímco mikrosatelitní často méně než 100 párů bazí. Ačkoliv většina polymorfismů je zřejmě funkčně neutrální, část z nich zřejmě má alelicky specifické účinky na regulaci genové exprese nebo funkce kódovaného proteinu, což determinuje interindividuální variabilitu v biologických znacích i vnímavost vůči nemoci.

14
Komplexní (multifaktoriální, multigenní) nemoci
Za genetickou predispozici mnoha biologických procesů, evolučních adaptací a tedy také tzv. komplexních nemocí zřejmě odpovídají kombinace určitých genů a určitých faktorů zevního prostředí. Interakční efekty a vliv vnějších faktorů však nutně musíme očekávat i v případě mendelisticky děděných nemocí, což se koneckonců projevuje ve všeobecně známé lékařské zkušenosti se širším klinickým spektrem příznaků stejného onemocnění.

15
Komplexní (multifaktoriální, multigenní) nemoci
Na odhalení nejobecnějších principů genetiky multifaktoriálních nemocí se na rozdíl od genetiky nemocí mendelistických v současné době stále ještě čeká. Také z tohoto důvodu zatím v klinické praxi často kolísá názor na výsledky genetických studií, které se snaží odhalit genetický podklad komplexních nemocí, od neodůvodněného očekávání nad nalezenými geny velkého účinku až po velkou skepsi vzhledem k existenci genetického podkladu v populaci četných nemocí ( nad 1%), jako je v kardiologii např. esenciální hypertenze. Jisté je, že pokud choroba má prokazatelně familiární výskyt, musíme očekávat podíl genetického podkladu na její manifestaci, a to i v tom případě, že není dosud dobře definován nebo dosavadní znalost nepovažujeme za přesvědčivou.
, jako je v kardiologii např. esenciální hypertenze. Jisté je, že pokud choroba má prokazatelně familiární výskyt, musíme očekávat podíl genetického podkladu na její manifestaci, a to i v tom případě, že není dosud dobře definován nebo dosavadní znalost nepovažujeme za přesvědčivou.")
16
Komplexní (multifaktoriální, multigenní) nemoci
Jinak řečeno, v 21. století již musíme počítat s tím, že fakticky každá choroba má nějaké genetické pozadí, jehož podíl na manifestaci dané choroby je různý. Své genetické pozadí mají i tak relativně vzdálené proximální fenotypy (viz níže), jako je např. kvalita života u nemocných s chronickým kardiovaskulárním onemocněním.
, jako je např. kvalita života u nemocných s chronickým kardiovaskulárním onemocněním.")
17
Genetické studie Základní debata nad genetickým podkladem nemocí logicky začíná od strategie výběru kandidátních genů. Tato otázka je podstatně jednodušší u mendelisticky děděných nemocí, kde se změněná funkce jednoho genu snadněji identifikuje. Dalším významným momentem je výběr statistické metodologie, která zhodnotí sílu asociace genů s chorobami. Možnosti jsou v zásadě dvě: linkage (vazebná) analýza a asociační studie. K detekci specifických genetických oblastí a genů, které se účastní v transmisi nemoci, je v principu možné použít obě metody.
 analýza a asociační studie. K detekci specifických genetických oblastí a genů, které se účastní v transmisi nemoci, je v principu možné použít obě metody.")
18
Genetické studie Asociační studie vyšetřují souvýskyt markeru a nemoci na populační úrovni, tj. u nepříbuzných jedinců, obvykle srovnáním frekvencí markerů u nepříbuzných nemocných a kontrolních subjektů (studie case-control). Statistickou sílu asociace je možno dále zvýšit obohacením o další kritéria, jako jsou klinické subtypy nemoci (studie case-case), závažnost nemoci, časný začátek nemoci, rizikové faktory pro nemoc včetně pohlaví a vhodné biologické znaky (např. plasmatické hladiny cytokinů při asociaci genetických polymorfismů v cytokinových genech; studie genotyp-fenotyp).
. Statistickou sílu asociace je možno dále zvýšit obohacením o další kritéria, jako jsou klinické subtypy nemoci (studie case-case), závažnost nemoci, časný začátek nemoci, rizikové faktory pro nemoc včetně pohlaví a vhodné biologické znaky (např. plasmatické hladiny cytokinů při asociaci genetických polymorfismů v cytokinových genech; studie genotyp-fenotyp).")
19
Kandidátní geny - asociace
s intermediálním fenotypem s klinickou manifestací nemoci s klinickou závažností nemoci s odpovídavostí nemoci na léčbu (účinnost, vedlejší příznaky)
")
20
DNA markery U komplexních nemocí se ukazuje, že je možno asociovat alely mnohých polymorfismů s výskytem komplexní nemoci nebo některými intermediálními znaky onemocnění (hladiny proteinů, rodinná anamnéza aj.) statisticky asociovat, čili přinejmenším najít genetický marker, s touto nemocí asociovaný. Určitý genotyp nebo alela daného polymorfismu tak představuje vyšší (nižší) riziko pro nemoc. Odds ratio (OR): Počet nemocných s riz. genotypem x počet zdravých bez riz. genotypu Počet nemocných bez riz. genotypu x počet zdravých s riz. genotypem
 statisticky asociovat, čili přinejmenším najít genetický marker, s touto nemocí asociovaný. Určitý genotyp nebo alela daného polymorfismu tak představuje vyšší (nižší) riziko pro nemoc. Odds ratio (OR): Počet nemocných s riz. genotypem x počet zdravých bez riz. genotypu. Počet nemocných bez riz. genotypu x počet zdravých s riz. genotypem.")
21
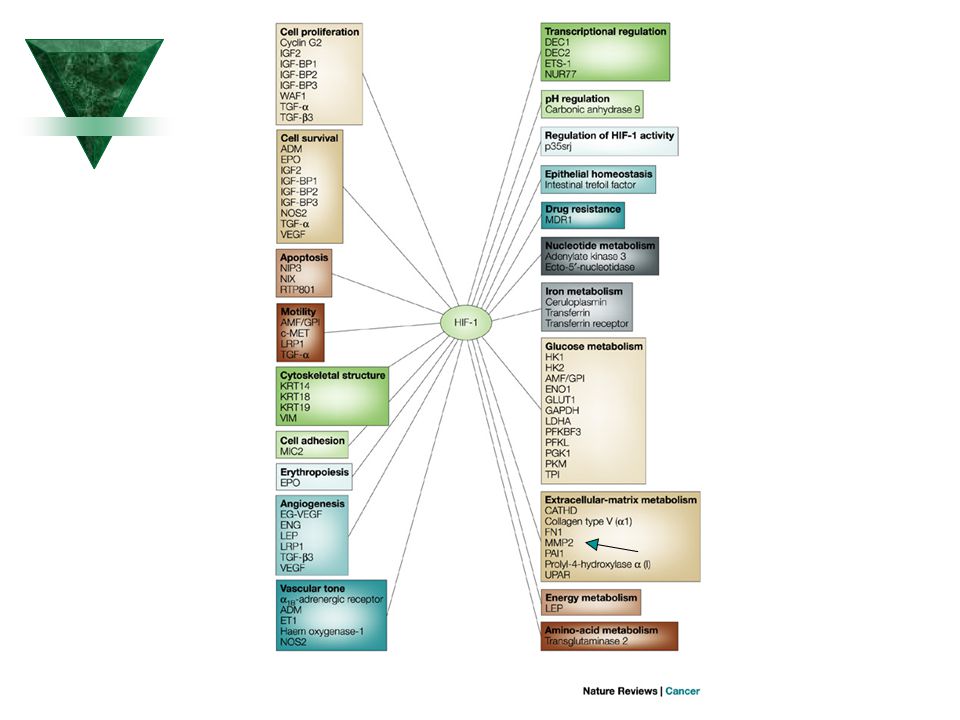
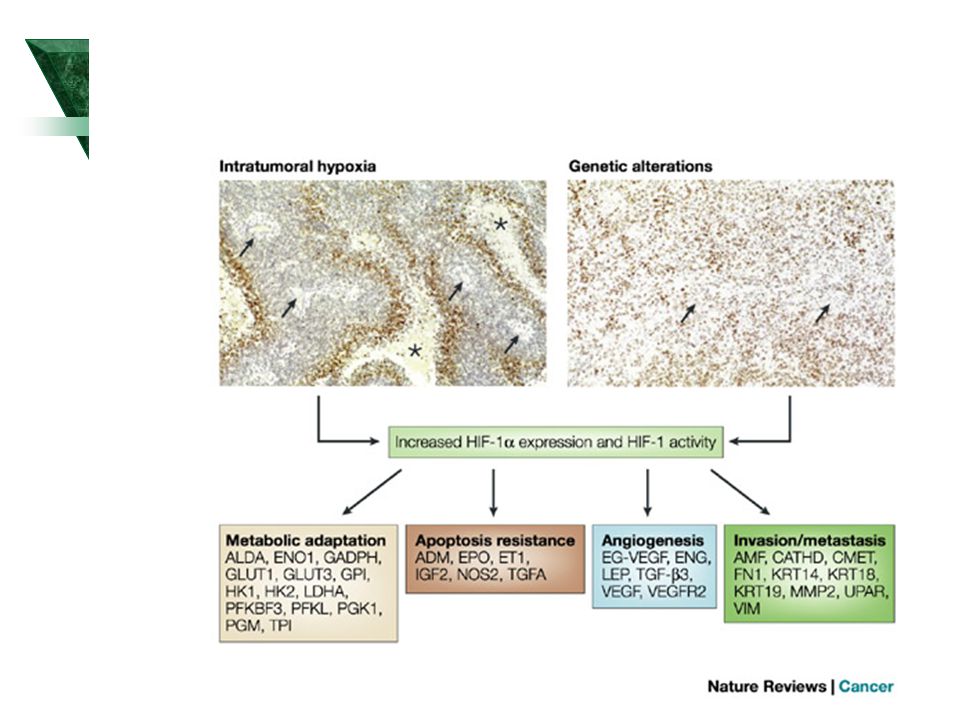
Výběr kandidátních genů pro komplexní nemoci
Základní fyziologické/patofyziologické děje: Hypoxie Hypoglykémie Zánět

23
HIF1a Pathway 23 ProteinLounge.com Glucose Glucose Glucose Transporter
2009 ProteinLounge.com C Glucose Transporter Glucose Transporter MKK6 MKK3 HSP70 P TCP1a P Normoxia Elongin-B VHL VHL Cytoplasmic Storage Vesicle Elongin-C P Hypoxia ERK2 PHD2 Elongin-B PHD1 VHL Ref1 Elongin-C FIH1 Cul2 Ub RBX1 Glucose Transporters OH Ub HIF1a Ub Ub Ub HSP90 HIF1a HIF1a OH PSM-a7 Proteasome Ub P LDHA HIF1a MDM2 p53 p53 Hypoxia Pathway Review: The cellular response to O2 (oxygen) is a central process in animal cells and figures prominently in the pathophysiology of several diseases, including cancer, cardiovascular disease, and stroke. This process is coordinated by the HIF (Hypoxia-Inducible Factor) and its regulator, the pVHL (Von Hippel-Lindau tumor suppressor protein). HIF1 is a basic helix-loop-helix transcription factor that transactivates genes encoding proteins that participate in homeostatic responses to hypoxia. It induces expression of proteins controlling glucose metabolism, cell proliferation, and vascularization. Several genes involved in cellular differentiation are directly or indirectly regulated by hypoxia. These include Epo (Erythropoietin), LDHA (Lactate Dehydrogenase-A), ET1 (Endothelin-1), transferrin, transferrin receptor, VEGF (Vascular Endothelial Growth Factor), Flk1, FLT1 (Fms-Related Tyrosine Kinase-1), PDGF-Beta (Platelet-Derived Growth Factor-Beta), bFGF (basic Fibroblast Growth Factor), and others genes affecting glycolysis (Ref.1). HIF1 consists of a heterodimer of two basic helix-loop-helix PAS (Per-ARNT-Sim) proteins, HIF1-Alpha, and HIF1-Beta. HIF1-Alpha accumulates under hypoxic conditions whereas HIF1-Beta is constitutively expressed. HIF1-Alpha is an important mediator of the hypoxic response of tumor cells and controls the up-regulation of a number of factors important for solid tumor expansion including the angiogenic factor VEGF. HIF1-Beta is the ARNT (Aryl hydrocarbon Receptor Nuclear Translocator), an essential component of the xenobiotic response (Ref.2). In the presence of O2, HIF is targeted for destruction by an E3 ubiquitin ligase containing the pVHL. Human pVHL binds to a short HIF-derived peptide when a conserved proline residue at the core of this peptide is hydroxylated. The human genome contains EGL9 (Egg Laying Nine-9) homologues that are named EGLN1, EGLN2, and EGLN3 (also called PHD2, PHD1, and PHD3 (Prolyl Hydroxylase Domain–Containing Proteins) respectively). Prolyl hydroxylase post-translationally modifies HIF1-Alpha, allowing it to interact with the VHL complex. Prolyl hydroxylase contains an iron moiety, so iron chelation inhibits this activity. All three proteins of Prolyl hydroxylase can hydroxylate HIF1-Alpha at one of two proline sites within the ODD (Pro-402 and Pro-564). Analogous prolyl residues are present in HIF2-Alpha and HIF3-Alpha. In the presence of oxygen, the EGLN proteins are active and hydroxylate the ODD domain of HIF1-Alpha, which allows pVHL to bind and polyubiquitinate HIF (Ref.3). VHL is part of a larger complex that includes Elongin-B, Elongin-C, Cul2, RBX1 (Ring-Box 1) and a ubiquitin-conjugating enzyme (E2). This complex, together with a ubiquitin-activating enzyme (E1), mediates the Ub (Ubiquitylation) of HIF1-Alpha. The Ub modification targets HIF1-Alpha for degradation, which can be blocked by proteasome inhibitors. Under hypoxic conditions the HIF1-Alpha subunits are not recognized by pVHL, and they consequently accumulate and dimerize with HIF1-Beta and translocates to the nucleus, where they interacts with cofactors such as CBP (CREB Binding Protein)/p300 and the Pol II (DNA polymerase II) complex to bind to HREs (Hypoxia-Responsive Element) and activate transcription of target genes. HIF1-Alpha-activated genes include VEGF, which promotes angiogenesis; GLUT1 (Glucose Transporter-1), which activates glucose transport; LDHA (Lactate Dehydrogenase), which is involved in the glycolytic pathway; and Epo, which induces erythropoiesis. HIF1-Alpha also activates transcription of NOS (Nitric Oxide Synthase), which promotes angiogenesis and vasodilation. ARNT2 and MOP3 (Member of Pas superfamily-3) are other proteins that have been shown to heterodimerize with HIF1-Alpha (Ref.4). HIF1-Alpha can also be regulated by ERK2, which phosphorylate HIF1-Alpha. HIF1-Alpha also associates with the molecular chaperone HSP90 (Heat Shock Protein-90). HSP90 antagonists also inhibited HIF1-Alpha transcriptional activity and dramatically reduced both hypoxia-induced accumulation of VEGF mRNA and hypoxia-dependent angiogenic activity. Recently, a factor inhibiting HIF1-Alpha activation, FIH (Factor Inhibiting HIF1-Alpha), has been described, representing a further level of HIF regulation. Hypoxia also induces p53 protein accumulation. p53 directly interacts with HIF1-Alpha and limits hypoxia-induced expression of HIF1-Alpha by promoting MDM2-mediated ubiquitination and proteasomal degradation under hypoxic conditions. Furthermore, the degradation of HIF1-Alpha by p53 in a hypoxic condition is inhibited by direct interaction with the JAB1 (Jun Activation domain Binding protein-1) and the ODD domain by blocking the interaction with p53. HIF1-Alpha also associates with HNF4alpha2 (Hepatocyte Nuclear Factor-4-Alpha 2), which activates the Epo gene in concert with HIF1-Alpha in response to hypoxic conditions. Hypoxia contributes significantly to the pathophysiology of major categories of human disease, including myocardial and cerebral ischemia, cancer, pulmonary hypertension, congenital heart disease and chronic obstructive pulmonary diseases (Ref.5). References: 1. Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem Aug 1;272(31): PubMed ID: 2. John F. O'Rourke, Ya-Min Tian, Peter J. Ratcliffe, and Christopher W. Pugh Oxygen-regulated and transactivating domains in endothelial PAS protein 1: comparison with hypoxia-inducible factor-1alpha. J. Biol. Chem Jan 22;274(4): PubMed ID: 3. Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J Sep 17;20(18): PubMed ID: 4. Damert A, Ikeda E, Risau W. Activator-protein-1 binding potentiates the hypoxia-induciblefactor-1-mediated hypoxia-induced transcriptional activation of vascular-endothelial growth factor expression in C6 glioma cells. Biochem. J Oct 15;327 ( Pt 2): PubMed ID: 5. Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol. Med Aug;7(8):345-50 PubMed ID: ARNT HIF1a Degradation ATP Pyruvate Per1 JAB1 ET1 HIF1a HNF4a2 Asparagine Hydroxylase Epo p16(INK4A) SRC1 Glucose HIF1a VEGF ARNT HIF1a VEGF Pathway CBP CREB Gene Expression NOSIII p300 c-Jun HRE NOS Pathway 23
 is a central process in animal cells and figures prominently in the pathophysiology of several diseases, including cancer, cardiovascular disease, and stroke. This process is coordinated by the HIF (Hypoxia-Inducible Factor) and its regulator, the pVHL (Von Hippel-Lindau tumor suppressor protein). HIF1 is a basic helix-loop-helix transcription factor that transactivates genes encoding proteins that participate in homeostatic responses to hypoxia. It induces expression of proteins controlling glucose metabolism, cell proliferation, and vascularization. Several genes involved in cellular differentiation are directly or indirectly regulated by hypoxia. These include Epo (Erythropoietin), LDHA (Lactate Dehydrogenase-A), ET1 (Endothelin-1), transferrin, transferrin receptor, VEGF (Vascular Endothelial Growth Factor), Flk1, FLT1 (Fms-Related Tyrosine Kinase-1), PDGF-Beta (Platelet-Derived Growth Factor-Beta), bFGF (basic Fibroblast Growth Factor), and others genes affecting glycolysis (Ref.1). HIF1 consists of a heterodimer of two basic helix-loop-helix PAS (Per-ARNT-Sim) proteins, HIF1-Alpha, and HIF1-Beta. HIF1-Alpha accumulates under hypoxic conditions whereas HIF1-Beta is constitutively expressed. HIF1-Alpha is an important mediator of the hypoxic response of tumor cells and controls the up-regulation of a number of factors important for solid tumor expansion including the angiogenic factor VEGF. HIF1-Beta is the ARNT (Aryl hydrocarbon Receptor Nuclear Translocator), an essential component of the xenobiotic response (Ref.2). In the presence of O2, HIF is targeted for destruction by an E3 ubiquitin ligase containing the pVHL. Human pVHL binds to a short HIF-derived peptide when a conserved proline residue at the core of this peptide is hydroxylated. The human genome contains EGL9 (Egg Laying Nine-9) homologues that are named EGLN1, EGLN2, and EGLN3 (also called PHD2, PHD1, and PHD3 (Prolyl Hydroxylase Domain–Containing Proteins) respectively). Prolyl hydroxylase post-translationally modifies HIF1-Alpha, allowing it to interact with the VHL complex. Prolyl hydroxylase contains an iron moiety, so iron chelation inhibits this activity. All three proteins of Prolyl hydroxylase can hydroxylate HIF1-Alpha at one of two proline sites within the ODD (Pro-402 and Pro-564). Analogous prolyl residues are present in HIF2-Alpha and HIF3-Alpha. In the presence of oxygen, the EGLN proteins are active and hydroxylate the ODD domain of HIF1-Alpha, which allows pVHL to bind and polyubiquitinate HIF (Ref.3). VHL is part of a larger complex that includes Elongin-B, Elongin-C, Cul2, RBX1 (Ring-Box 1) and a ubiquitin-conjugating enzyme (E2). This complex, together with a ubiquitin-activating enzyme (E1), mediates the Ub (Ubiquitylation) of HIF1-Alpha. The Ub modification targets HIF1-Alpha for degradation, which can be blocked by proteasome inhibitors. Under hypoxic conditions the HIF1-Alpha subunits are not recognized by pVHL, and they consequently accumulate and dimerize with HIF1-Beta and translocates to the nucleus, where they interacts with cofactors such as CBP (CREB Binding Protein)/p300 and the Pol II (DNA polymerase II) complex to bind to HREs (Hypoxia-Responsive Element) and activate transcription of target genes. HIF1-Alpha-activated genes include VEGF, which promotes angiogenesis; GLUT1 (Glucose Transporter-1), which activates glucose transport; LDHA (Lactate Dehydrogenase), which is involved in the glycolytic pathway; and Epo, which induces erythropoiesis. HIF1-Alpha also activates transcription of NOS (Nitric Oxide Synthase), which promotes angiogenesis and vasodilation. ARNT2 and MOP3 (Member of Pas superfamily-3) are other proteins that have been shown to heterodimerize with HIF1-Alpha (Ref.4). HIF1-Alpha can also be regulated by ERK2, which phosphorylate HIF1-Alpha. HIF1-Alpha also associates with the molecular chaperone HSP90 (Heat Shock Protein-90). HSP90 antagonists also inhibited HIF1-Alpha transcriptional activity and dramatically reduced both hypoxia-induced accumulation of VEGF mRNA and hypoxia-dependent angiogenic activity. Recently, a factor inhibiting HIF1-Alpha activation, FIH (Factor Inhibiting HIF1-Alpha), has been described, representing a further level of HIF regulation. Hypoxia also induces p53 protein accumulation. p53 directly interacts with HIF1-Alpha and limits hypoxia-induced expression of HIF1-Alpha by promoting MDM2-mediated ubiquitination and proteasomal degradation under hypoxic conditions. Furthermore, the degradation of HIF1-Alpha by p53 in a hypoxic condition is inhibited by direct interaction with the JAB1 (Jun Activation domain Binding protein-1) and the ODD domain by blocking the interaction with p53. HIF1-Alpha also associates with HNF4alpha2 (Hepatocyte Nuclear Factor-4-Alpha 2), which activates the Epo gene in concert with HIF1-Alpha in response to hypoxic conditions. Hypoxia contributes significantly to the pathophysiology of major categories of human disease, including myocardial and cerebral ischemia, cancer, pulmonary hypertension, congenital heart disease and chronic obstructive pulmonary diseases (Ref.5). References: 1. Jiang BH, Zheng JZ, Leung SW, Roe R, Semenza GL. Transactivation and inhibitory domains of hypoxia-inducible factor 1alpha. Modulation of transcriptional activity by oxygen tension. J. Biol. Chem Aug 1;272(31): PubMed ID: John F. O Rourke, Ya-Min Tian, Peter J. Ratcliffe, and Christopher W. Pugh. Oxygen-regulated and transactivating domains in endothelial PAS protein 1: comparison with hypoxia-inducible factor-1alpha. J. Biol. Chem Jan 22;274(4): PubMed ID: Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ. Independent function of two destruction domains in hypoxia-inducible factor-alpha chains activated by prolyl hydroxylation. EMBO J Sep 17;20(18): PubMed ID: Damert A, Ikeda E, Risau W. Activator-protein-1 binding potentiates the hypoxia-induciblefactor-1-mediated hypoxia-induced transcriptional activation of vascular-endothelial growth factor expression in C6 glioma cells. Biochem. J Oct 15;327 ( Pt 2): PubMed ID: Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol. Med Aug;7(8): PubMed ID: ARNT. HIF1a. Degradation. ATP. Pyruvate. Per1. JAB1. ET1. HIF1a. HNF4a2. Asparagine. Hydroxylase. Epo. p16(INK4A) SRC1. Glucose. HIF1a. VEGF. ARNT. HIF1a. VEGF. Pathway. CBP. CREB. Gene Expression. NOSIII. p300. c-Jun. HRE. NOS Pathway. 23.")
25
Chow et al., 2007

26
Clark, I. M., et al., The regulation of matrix metalloproteinases and their inhibitors, Int J
Biochem Cell Biol (2008), doi: /j.biocel
, doi: /j.biocel")
27
Clark, I. M., et al., The regulation of matrix metalloproteinases and their inhibitors, Int J
Biochem Cell Biol (2008), doi: /j.biocel
, doi: /j.biocel")
28
Chow et al., 2007

29
Cíle pro MMP-2 po poškození typu ischémie/reperfúze Chow et al., 2007
Kardiomyocyty Myosin light chain Cíle pro MMP-2 po poškození typu ischémie/reperfúze Chow et al., 2007

30
Chow et al., 2007

31
William Hornebeck and François Xavier Maquart, 2003
RGD=LEUCINE-RICH REPEAT-, TROPOMODULIN DOMAIN-, AND PROLINE-RICH DOMAIN-CONTAINING PROTEIN William Hornebeck and François Xavier Maquart, 2003

32
Kolagenem (-y) řízený β1–β3 „switch“ a regulace buněčné migrace
Kolagen typu IV: akce MMP-2, MMP-9 (nebo jiných enzymů) odkrývá místa na kolagenu typu IV, která reagují s vβ3 a podporují buněčnou migraci. Extenzivnější proteolýza vede k uvolnění peptidu 3CB IV, který inhibuje prostřednictvím interakce s β3 buněčný růst a migraci (down-regulation MT1-MMP a vβ3). Kolagen typu I: interakce řízená 2β1 mezi tímto typem kolagenu a buňkami melanomu indukuje expresi MMP-1, MT1-MMP a aktivaci MMP-2; tyto enzymy mohou spolupracovat na degradaci kolagenu.
 odkrývá místa na kolagenu typu IV, která reagují s vβ3 a podporují buněčnou migraci. Extenzivnější proteolýza vede k uvolnění peptidu 3CB IV, který inhibuje prostřednictvím interakce s β3 buněčný růst a migraci (down-regulation MT1-MMP a vβ3). Kolagen typu I: interakce řízená 2β1 mezi tímto typem kolagenu a buňkami melanomu indukuje expresi MMP-1, MT1-MMP a aktivaci MMP-2; tyto enzymy mohou spolupracovat na degradaci kolagenu.")
33
Gen pro MMP2 – lokalizace 16q13
Promotor genu -735 C T 0.12

34
MMP-2 (-1306 A/G) Tranzice C --> T v pozici (alelická frekvence 0.26), která mění promotorové místo Sp1-typu (CCACC box), způsobuje nižší promotorovou aktivitu alely T (Price et al., 2001) p16 oslabuje Sp1 vazbu na promotor MMP-2 a suprimuje transkripci genu. Zvýšená exprese Sp1 může hrát proti této p16-indukované snížené expresi MMP-2. Komplex cyklinA/CDKináza může přímo fosforylovat Sp1 a podporovat jeho vaznou aktivitu na DNA (Wang et al., 2006)
, která mění promotorové místo Sp1-typu (CCACC box), způsobuje nižší promotorovou aktivitu alely T (Price et al., 2001) p16 oslabuje Sp1 vazbu na promotor MMP-2 a suprimuje transkripci genu. Zvýšená exprese Sp1 může hrát proti této p16-indukované snížené expresi MMP-2. Komplex cyklinA/CDKináza může přímo fosforylovat Sp1 a podporovat jeho vaznou aktivitu na DNA (Wang et al., 2006)")
36
MMP-2 (-790 T/G) Alela T, nikoliv G obsahuje sekvenci 3 transkripčních faktorů: GKLF (gut-enriched Krueppel-like factor) Účastní se proliferace a diferenciace tkání Knockoutované myši těžké malformace str. corneum Účast v patogeneze mnohočetného myelomu (zvýšeně exprimován při blokádě receptoru 3 pro fibroblastový růstový faktor in vitro) Umlčován v buňkách „adult T-cell leukemia“ S8 Povrchový antigen Evi1(ectopic viral integration site 1 encoded factor) Inhibuje signální dráhu cytokinů rodiny TGF a tím i apoptózu navozovanou TGF změněná (zvýšená) exprese u lidských myelodysplastických syndromů a jiných hematologických malignit
 Umlčován v buňkách „adult T-cell leukemia S8. Povrchový antigen. Evi1(ectopic viral integration site 1 encoded factor) Inhibuje signální dráhu cytokinů rodiny TGF a tím i apoptózu navozovanou TGF změněná (zvýšená) exprese u lidských myelodysplastických syndromů a jiných hematologických malignit.")
37
MMP-2 a nemoci kardiovaskulární MMP-2-1575 G/A
NS

38
MMP-2 a nemoci kardiovaskulární MMP-2 -1306 T/C
NS

39
MMP-2 a nemoci kardiovaskulární MMP-2 -790 G/T
Pg=0,01, Pa=0,03 Pg=0,007, Pa=0,009 Pg=0,02, Pa=0,009 Pg=0,01, Pa=0,02

40
MMP-2 a nemoci kardiovaskulární MMP-2 -735 T/C
NS

41
MMP-2 a nemoci kardiovaskulární Chronické srdeční selhání a celkový cholesterol
ATGT : OR=14,3; P=0,00075 – Pcorr=0,006 Síla testu– 85% při = 5% Dostatečná velikost souboru

42
MMP-2 a nemoci kardiovaskulární DILATAČNÍ KARDIOMYOPATIE MMP2 a celkový cholesterol
GCTT: OR=8,75, P=0,01 pro DKMP s vysokou hladinou cholesterolu ATGT: OR=0,12, P=0,03 pro DKMP s vysokou hladinou cholesterolu

43
MMP-2 a nemoci kardiovaskulární DILATAČNÍ KARDIOMYOPATIE VS
MMP-2 a nemoci kardiovaskulární DILATAČNÍ KARDIOMYOPATIE VS. ISCHEMICKÁ CHOROBA SRDEČNÍ MMP2 a celkový cholesterol pod 5 mmol/l GCTT: OR=11, P=0,002 pro ICHS oproti DKMP, 80% síla testu na = 5% ATGT: OR=11,92, P=0,008 pro DKMP oproti ICHS, 65% síla testu na = 5%

44
MMP-2 a nemoci kardiovaskulární CHRONICKÉ SRDEČNÍ SELHÁNÍ MMP2 a LDL
GCTC : P=0,02

45
MMP-2 a nemoci kardiovaskulární
Haplotyp ATGT čtyř promotorových polymorfismů v genu pro MMP-2 nese vysoké riziko (OR=14, Pcorr=0,006) pro pacienty chronickým srdečním selháním a nízkou hladinou cholesterolu oproti pacientům s vysokou hladinou cholesterolu. Haplotyp ATGT nese vysoké riziko (OR=9, P=0,01) pro pacienty s dilatační kardiomyopatií a nízkou hladinou cholesterolu oproti pacientům s vysokou hladinou cholesterolu. Neprokázali jsme asociaci žádného haplotypu v promotoru genu pro MMP-2 s ischemickou chorobou srdeční a nízkou nebo vysokou hladinou celkového cholesterolu.
 pro pacienty chronickým srdečním selháním a nízkou hladinou cholesterolu oproti pacientům s vysokou hladinou cholesterolu. Haplotyp ATGT nese vysoké riziko (OR=9, P=0,01) pro pacienty s dilatační kardiomyopatií a nízkou hladinou cholesterolu oproti pacientům s vysokou hladinou cholesterolu. Neprokázali jsme asociaci žádného haplotypu v promotoru genu pro MMP-2 s ischemickou chorobou srdeční a nízkou nebo vysokou hladinou celkového cholesterolu.")
46
MMP-2 a nemoci kardiovaskulární
Haplotyp ATGT nese zvýšené riziko (OR=12, P=0,008) pro srdečně selhávající pacienty s dilatační kardiomyopatii oproti pacientům s ischemickou chorobou srdeční. Haplotyp GCTT nese naopak vyšší riziko (OR=11, P=0,002) pro pacienty s ICHS oproti srdečně selhávajícím pacientům s dilatační kardiomyopatií. Haplotyp četných promotorových polymorfismů jsme asociovali pouze s celkovou hladinou cholesterolu, nikoliv s dalšími parametry lipidového metabolismu.
 pro srdečně selhávající pacienty s dilatační kardiomyopatii oproti pacientům s ischemickou chorobou srdeční. Haplotyp GCTT nese naopak vyšší riziko (OR=11, P=0,002) pro pacienty s ICHS oproti srdečně selhávajícím pacientům s dilatační kardiomyopatií. Haplotyp četných promotorových polymorfismů jsme asociovali pouze s celkovou hladinou cholesterolu, nikoliv s dalšími parametry lipidového metabolismu.")
47
MMP-2 a kožní T-lymfomy Pg=0,06, Pa=0,04, odds ratio pro CC u MF, SS=3,29, Pcorr=0,05

48
MMP-2 a kožní T-lymfomy Pg=0,01, Pa=0,009, odds ratio pro TT u MF, SS=3,25, Pcorr=0,05

49
MMP-2 a kožní T-lymfomy Pg=0,02, Pa=0,008, odds ratio pro GG u MF Ia=4,80, Pcorr=0,03

50
MMP-2 a kožní T-lymfomy Pg=0,009, Pa=0,007, odds ratio pro CC u MF Ia=7,31, Pcorr=0,008

51
MMP-2 a kožní T-lymfomy Pg=0,007, Pa=0,003, odds ratio pro TT u MF Ia=7,77, Pcorr=0,006

52
MMP-2 a kožní T-lymfomy

53
Phillips MI et al., 2008

54
Patofyziologické souvislosti mezi genetickou variabilitou v ATG
Reakce akutní fáze a ateroskleróza? Reakce akutní fáze a rakovina? Reakce akutní fáze a autoimunita?

55
Frekvence 7 haplotypových skupin v genu pro ATG definovaných 19 polymorfismy
-1074 -812 -792 -775 -532 -217 -6 68 172 384 409 507 676 698 1164 2186 4072 5093 5593 Frequency Percentage H1 T C A G 17 16.3 H2 12 11.5 H3 8 7.7 H4 21 20.2 H5 7 6.73 H6 30 28.8 H7 9 8.7 Chimpanzee Fejerman L et al., 2004

56
Párové dysekvilibrium (D ) pro angiotenzinogen
Párové dysekvilibrium (D ) pro angiotenzinogen. Rozsah D 0,01 to 1,00, od tvavomodré po červenou. Horizontální a vertikální osy vyjadřují fyzikální vzdálenosti mezi markery. Fejerman L et al., 2004
 pro angiotenzinogen. Rozsah D 0,01 to 1,00, od tvavomodré po červenou. Horizontální a vertikální osy vyjadřují fyzikální vzdálenosti mezi markery. Fejerman L et al.,")
57
„Haplogroup tree“ pro gen pro angiotenzinogen
„Haplogroup tree“ pro gen pro angiotenzinogen. Každý kruh reprezentuje haploskupinu. N je počet chromosomů v každé skupině a velikost kruhu reflektuje velikost N. Délka ramen je proporcionální počtu mutací(z 19 SNP vybraných pro definici haplotypových skupin), které oddělují haplotypové skupiny. Hvězdičky jsou mutace. Černé hvězdičky pro totožné nukleotidy se šimpanzy. Fejerman L et al., 2004
, které oddělují haplotypové skupiny. Hvězdičky jsou mutace. Černé hvězdičky pro totožné nukleotidy se šimpanzy. Fejerman L et al.,")
58
Haplotypy pro AGT, založené na 21 SNP a šimpanzí sekvenci
Haplotypy pro AGT, založené na 21 SNP a šimpanzí sekvenci. Velikost kruhů odráží frekvence haplotypů u Kaukaziánů (A) a Japonců (B). Am J Hum Genet January; 70(1): 108–123. Published online 2001 November 30.
 a Japonců (B). Am J Hum Genet January; 70(1): 108–123. Published online 2001 November 30.")
59
Struktura genu pro AGT: a G-1074T, b C-532T, c G-217A, d A-20C, e A-6G, f C68T, g C3889T (T174M), h C4072T (T235M).
, h C4072T (T235M).")
60
Angiotensinogen a nemoci kardiovaskulární M235T ATG
Pg = 0,06

61
Angiotensinogen a nemoci kardiovaskulární ATG-6A/G
Pg=0,001 Pg=0,07

62
Příklady asociačních studií: Haplotypy- -6A/G a M235T ATG
OR=7,15, Pcorr=0,00001, ST =99%-98,5%. OR=5,75, Pcorr=0,01, ST =95%-83%.

63
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS (ICHS) vs
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS (ICHS) vs. pacienti s CAD (3K) Haplotypy ATG (-6A/G + M235T) Vyšší frekvence výskytu GT haplotypu u pacientů s CHSS pro ICHS OR = 6,35; Pcorr. = 0,0006, PT- 95%
 vs. pacienti s CAD (3K) Haplotypy ATG (-6A/G + M235T) Vyšší frekvence výskytu GT haplotypu u pacientů s CHSS pro ICHS. OR = 6,35; Pcorr. = 0,0006, PT- 95%")
64
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS (DKM) vs
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS (DKM) vs. pacienti s CAD (3K) Haplotypy ATG (-6A/G + M235T) Vyšší frekvence výskytu GT haplotypu u pacientů s CHSS pro DKM OR = 7,86; Pcorr = 0,0001 PT= 98%
 vs. pacienti s CAD (3K) Haplotypy ATG (-6A/G + M235T) Vyšší frekvence výskytu GT haplotypu u pacientů s CHSS pro DKM. OR = 7,86; Pcorr = 0,0001. PT= 98%")
65
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS vs
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS vs. kontroly Dvojgenotyp ATG (-6A/G a M235T ATG OR=2,63 (95% konfidenční interval 1,39-4,95), P=0,002, Pcorr=0,02
, P=0,002, Pcorr=0,02.")
66
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS vs
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS vs. kontroly-ženy Dvojgenotyp ATG (-6A/G a M235T ATG) OR=15,5 (95% konfidenční interval 1,86-129,42), P=0,002, Pcorr=0,008
 OR=15,5 (95% konfidenční interval 1,86-129,42), P=0,002, Pcorr=0,008.")
67
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS vs
Angiotensinogen a nemoci kardiovaskulární Pacienti s CHSS vs. pacienti s CAD (3K) Haplotypy ATG (-6A/G a M235T ATG) OR=7,15, Pcorr=0,00001, PT =99%-98,5%. OR=0,17, Pcorr=0,01, PT =95%-83%.
 Haplotypy ATG (-6A/G a M235T ATG) OR=7,15, Pcorr=0,00001, PT =99%-98,5%. OR=0,17, Pcorr=0,01, PT =95%-83%.")
68
Angiotensinogen a nemoci kardiovaskulární Hypertonici vs
Angiotensinogen a nemoci kardiovaskulární Hypertonici vs. kontroly Alela A (-6A/G), alela M (M235T) ATG OR pro obézní hypertoniky =3,05, Pcorr=0,0001 OR pro obézní hypertoniky =1,95, Pcorr=0,02
, alela M (M235T) ATG. OR pro obézní hypertoniky. =3,05, Pcorr=0,0001. OR pro obézní hypertoniky. =1,95, Pcorr=0,02.")
69
Angiotensinogen a sclerosis multiplex
Pacienti se sclerosou multiplex vs. kontroly-ženy Dvojgenotyp ATG (-6A/G a M235T ATG) OR=17,65 (95% konfidenční interval 2,31-134,71; P= , Pcorr=0.0006)
 OR=17,65 (95% konfidenční interval 2,31-134,71; P= , Pcorr=0.0006)")
70
Angiotensinogen a sclerosis multiplex
Pacienti se sclerosou multiplex vs. kontroly-ženy Dvojgenotyp ATG (-6A/G a M235T ATG) OR=4,67 (95% konfidenční interval 1,31-16,63; P=0,008, Pcorr=0,04
 OR=4,67 (95% konfidenční interval 1,31-16,63; P=0,008, Pcorr=0,04.")
71
Angiotensinogen a sclerosis multiplex
Pacienti se sclerosou multiplex vs. kontroly-ženy Dvojgenotyp ATG (-6A/G a M235T ATG) OR=0,28 (95% konfidenční interval 0,13-0,61, P=0,0008, Pcorr=0,005
 OR=0,28 (95% konfidenční interval 0,13-0,61, P=0,0008, Pcorr=0,005.")
72
Vztah mezi zánětem a rakovinou

73
Patofyziologické mechanismy spojující obezitu a rakovinu střev
(John BJ, 2006) Patofyziologické mechanismy spojující obezitu a rakovinu střev
 Patofyziologické mechanismy spojující obezitu a rakovinu střev.")
74
Vztah mezi zánětem a rakovinou

75
Pacienti s kolorektální karcinomem vs
Pacienti s kolorektální karcinomem vs. kontroly-muži Dvojgenotyp ATG (-6A/G a M235T ATG) OR pro MTAG=3,82 (2,09-6,96) P=0,0002, Pcorr=0,002 OR pro MTGG=0,24 (0,08-0,75) P=0,007, Pcorr=0,05
 OR pro MTAG=3,82 (2,09-6,96) P=0,0002, Pcorr=0,002. OR pro MTGG=0,24 (0,08-0,75) P=0,007, Pcorr=0,05.")
76
Farmakogenetika a vývoj léků
Nutnost přesné diagnózy (k fenotypicky podobným stavům mohou vést různé patobiochemické mechanismy). Individuální odpověď jedince na terapii může záležet na genech, vstupujících do interakce s metabolismem léku nebo jeho působením. Polovina všech dosud používaných léků je metabolizována enzymy P450.
. Individuální odpověď jedince na terapii může záležet na genech, vstupujících do interakce s metabolismem léku nebo jeho působením. Polovina všech dosud používaných léků je metabolizována enzymy P450.")
77
Farmakologická léčba-241 pacientů s chronickým srdečním selháním
Inhibitory ACE A/N 241 (100%) Beta blokátory A/N 150/91 (62%) ACEI + BB A/N Diuretika A/N 214/27 (89%) Aspirin A/N 133/108(55%) Inhibitory Ca A/N 7/233 (3%) Verospiron A/N 157/84 (65%) Digoxin A/N 163/78 (68%) Inzulín A/N 20/221 (8%) Perorální antidiabetika A/N 46/195 (19%) Antikoagulační terapie A/N 52/189 (22%) Nitráty A/N 89/152 (37%) Milurit 57/184 (24%) Hypolipidemika 107/134 (44%)
 Beta blokátory A/N. 150/91 (62%) ACEI + BB A/N. Diuretika A/N. 214/27 (89%) Aspirin A/N. 133/108(55%) Inhibitory Ca A/N. 7/233 (3%) Verospiron A/N. 157/84 (65%) Digoxin A/N. 163/78 (68%) Inzulín A/N. 20/221 (8%) Perorální antidiabetika A/N. 46/195 (19%) Antikoagulační terapie A/N. 52/189 (22%) Nitráty A/N. 89/152 (37%) Milurit. 57/184 (24%) Hypolipidemika. 107/134 (44%)")
78
Detekce genotypů inzerčně delečního polymorfismu v genu
pro angiotenzin konvertující enzym (polymorfismus I/D ACE). PCR na horizontální elektroforéze ID DD II
. PCR na horizontální elektroforéze. ID. DD. II.")
79
Farmakogenetické rozdíly v dávkování beta blokátorů- genotyp I/D ACE
Pg=0,005, Pa=0,002

80
Farmakogenetické aspekty u pacientů s CHSS –polymorfismus I/D ACE
OR pro BB u II = 2,74, P=0.004 OR pro D u II = 7,76, P=0,01 OR pro Asp u II = 0,49, P=0,02

81
Farmakogenetické rozdíly v dávkování beta blokátorů a ACEI-polymorfismus I/D ACE
OR pro II u pacientů s nižší než poloviční dávkou BB a/nebo ACEI je 2,84, P=0,002 Pg=0,006, Pa=0,009

82
MR Stratton et al. Nature 458, 719-724 (2009) doi:10.1038/nature07943
 doi: /nature07943")
83
Děkuji vám za pozornost

Podobné prezentace