Stáhnout prezentaci
Prezentace se nahrává, počkejte prosím

1
Správná klinická praxe - legislativa
Zpracoval Doc. MUDr. Ivan Pohl, CSc. Leden 2012 Tato prezentace je spolufinancována Evropským sociálním fondem a státním rozpočtem České republiky

2
Správná klinická praxe z legislativního hlediska – léčiva (1)
Směrnice Evropského parlamentu a Rady 2001/20/ES (Clinical Trials Directive) Směrnice Komise 2005/28/ES ze dne 9. dubna 2005 (GCP Directive) Zákon č. 79/1997 Sb., o léčivech v platném znění Vyhláška MZd a MZ č. 226/2008 Sb., o správné klinické praxi a bližších podmínkách klinického hodnocení léčivých přípravků Tato prezentace je spolufinancována Evropským sociálním fondem a státním rozpočtem České republiky
 Směrnice Komise 2005/28/ES ze dne 9. dubna 2005 (GCP Directive) Zákon č. 79/1997 Sb., o léčivech v platném znění. Vyhláška MZd a MZ č. 226/2008 Sb., o správné klinické praxi a bližších podmínkách klinického hodnocení léčivých přípravků. Tato prezentace je spolufinancována Evropským sociálním fondem a státním rozpočtem České republiky.")
3
Správná klinická praxe z legislativního hlediska – léčiva (2)
trials/index_en.htm Směrnice Evropského parlamentu a Rady 2001/20/ES (Clinical Trials Directive) on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use Směrnice Komise 2005/28/ES ze dne 9. dubna (GCP Directive) laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products
 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Směrnice Komise 2005/28/ES ze dne 9. dubna 2005 (GCP Directive) laying down principles and detailed guidelines for good clinical practice as regards investigational medicinal products for human use, as well as the requirements for authorisation of the manufacturing or importation of such products.")
4
Správná klinická praxe z legislativního hlediska – léčiva (3)
Klinické zkoušky s léčivy provedené mimo EU Directive 2001/83/EC - Community code relating to medicinal products for human use Eudralex Volume 10 of the publications "The rules governing medicinal products in the European Union" contains guidance documents applying to clinical trials European Medicines Agency The outcome of the Agency's evaluation is used by the European Commission to decide whether a medicine can be authorised for marketing in the European Union (EU) EU Clinical Trials Register -
 EU Clinical Trials Register -")
5
Clinical Trials Directive (2001/20/EC) on approximation of laws, regulations … etc.
Sets standards for the protection of clinical trials subjects, including incapacitated adults and minors Requires Member States to establish ethics committees on a legal basis Covers certain Licensing Authority procedures for commencing a clinical trial Lays down standards for the manufacture, import and labelling of investigational medicinal products (IMPs) It also requires clinical trials to be conducted in compliance with the principles of good clinical practice (GCP)
 It also requires clinical trials to be conducted in compliance with the principles of good clinical practice (GCP)")
6
Good Clinical Practice Directive (2005/28/EC) adopted in April 2005
the principles of good clinical practice and detailed guidelines in line with those principles, the requirements for authorisation of the manufacture or importation of such products, the detailed guidelines on the documentation relating to clinical trials, archiving, qualifications of inspectors and inspection procedures.

7
Good Clinical Practice (GCP)
is an international ethical and scientific quality standard for designing, conducting, recording and reporting trials that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety and well-being of trial subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, and that the clinical trial data are credible.

8
Proč je správná klinická praxe důležitá?

9
Základní cíle správné klinické praxe
To protect the rights, safety and welfare of humans participating in research To assure the quality, reliability and integrity of data collected To provide standards and guidelines for the conduct of clinical research Good Clinical Practice = Ethics + Quality Data

10
GCP - 13 Principles Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirement(s). Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.
. Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.")
11
GCP - 13 Principles The rights, safety, and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society. The available nonclinical and clinical information on an investigational product should be adequate to support the proposed clinical trial. Clinical trials should be scientifically sound, and described in a clear, detailed protocol.

12
GCP - 13 Principles A trial should be conducted in compliance with the protocol that has received prior institutional review board (IRB)/independent ethics committee (IEC) approval/favourable opinion. Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s). The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified dentist.
/independent ethics committee (IEC) approval/favourable opinion. Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s). The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified dentist.")
13
GCP - 13 Principles Freely given informed consent should be obtained from every subject prior to clinical trial participation. All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation and verification. The confidentiality of records that could identify subjects should be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirement(s).
.")
14
GCP - 13 Principles Investigational products should be manufactured, handled, and stored in accordance with applicable good manufacturing practice (GMP). They should be used in accordance with the approved protocol. Systems with procedures that assure the quality of every aspect of the trial should be implemented.
. They should be used in accordance with the approved protocol. Systems with procedures that assure the quality of every aspect of the trial should be implemented.")
15
Vyhláška MZd a MZ č. 226/2008 Sb. Upravuje pravidla správné klinické praxe a bližší podmínky klinického hodnocení léčivých přípravků (dále jen „klinické hodnocení“).
.")
16
Správná klinická praxe
Soubor mezinárodně uznávaných etických a vědeckých požadavků na kvalitu, které musejí být dodrženy při navrhování klinických hodnocení s účastí lidských subjektů, při jejich provádění, dokumentování a při zpracování zpráv a hlášení o těchto hodnocení.

17
§ 10 Oznamování závažných nežádoucích příhod subjektu hodnocení
Vyhláška MZd ČR č. 226/2008 Sb. HLAVA PRVNÍ - Etická komise HLAVA DRUHÁ – Zkoušející § 7 Základní činnosti zkoušejícího § 8 Poučení a informovaný souhlas § 9 Záznamy a zprávy § 10 Oznamování závažných nežádoucích příhod subjektu hodnocení § 11 Přerušení klinického hodnocení a jeho ukončení

18
HLAVA TŘETÍ – Zadavatel
§ 12 Základní činnosti zadavatele § 13 Žádost o povolení a ohlášení klinického hodnocení Ústavu § 14 Vedení klinického hodnocení, sběr údajů a uchovávání záznamů § 15 Informace o průběhu klinického hodnocení § 16 Ohlášení změny podmínek klinického hodnocení § 17 Ostatní informace o hodnoceném léčivém přípravku a klinickém hodnocení § 18 Informace o ukončení klinického hodnocení a souhrnná zpráva § 19 Hodnocené léčivé přípravky a jejich označování § 20 Změna zadavatele

19
HLAVA ČTVRTÁ Monitorování a audit klinického hodnocení
Dohled nad klinickým hodnocením zajišťuje zadavatel monitorováním klinického hodnocení, kterým ověřuje především, že práva a bezpečnost všech subjektů hodnocení nejsou narušeny, zaznamenávané údaje jsou správné, úplné a ověřitelné na základě zdrojových dokumentů, klinické hodnocení probíhá v souladu s poslední schválenou verzí protokolu a jeho případnými dodatky správnou klinickou praxí a souvisejícími právními předpisy.

20
ČÁST TŘETÍ – Klinické hodnocení veterinárních léčivých přípravků
Příloha č. 1 k vyhlášce č. 226/2008 Sb. Protokol klinického hodnocení a dodatky k protokolu Příloha č. 2 k vyhlášce č. 226/2008 Sb. Poučení subjektu hodnocení a písemný informovaný souhlas

21
Protokol klinického hodnocení léčiv
Protokol musí mít obsah Na titulní straně musí být identifikační údaje, tj.: název a identifikační číslo protokolu jméno a adresa zadavatele/monitora oprávněná osoba zastupující zadavatele identifikace kvalifikovaného konzultanta (i telefon) identifikace zkoušejících identifikace zkoušejícího zodpovědného za lékařská rozhodnutí názvy, adresy zdravotnických zařízení účastnících se na klinických zkouškách, včetně laboratoří
 identifikace zkoušejících. identifikace zkoušejícího zodpovědného za lékařská rozhodnutí. názvy, adresy zdravotnických zařízení účastnících se na klinických zkouškách, včetně laboratoří.")
22
Obsah protokolu klinického hodnocení léčiv
Základní informace Cíle klinického hodnocení Plán klinického hodnocení Výběr subjektů a jejich vyřazení Léčba subjektů hodnocení Hodnocení účinnosti Statistika Přímý přístup ke zdrojovým dokumentům Zabezpečování a řízení jakosti Etické otázky Zacházení s údaji a uchovávání záznamů Financování a pojištění Zásady publikační činnosti

23
Poznámky k praktickému provádění vývoje léčiv, včetně klinických zkoušek/ aplikace SKP

24
Strnadová V. Farmaceutická medicína.

25
Strnadová V. Farmaceutická medicína.

26
Vývoj léku Preklinická fáze Klinická fáze Fáze I Fáze II Fáze III
Fáze IV

27
Preklinická fáze Primární famakologická aktivita Aplikace lékové formy
Farmakokinetika Účinek na tělesné orgány Toxikologie léku

28
Klinická fáze I Ověření účinku na jednotlivcích
Specializovaná klinická pracoviště Klinická fáze II První studie – stanovení vhodné dávky, cílové populace, statistický model Klinická fáze III Průkaz účinnosti na cílové populaci Stanovení profilu bezpečnosti

29
Klinická fáze IV Optimalizace použití po registraci ve schválené indikaci Průkaz méně obvyklých nežádoucích účinků Upřesnění dávkování Zařazení do léčebných standardů Farmakoekonomika

30
Správná klinická praxe v souvislosti s klinickým hodnocením / klinickými zkouškami zdravotnických prostředků

31
Zdravotnický prostředek
je nástroj, přístroj, zařízení, programové vybavení, materiál nebo jiný předmět, použitý samostatně nebo v kombinaci, spolu s příslušenstvím, včetně programového vybavení, určený výrobcem pro použití u člověka za účelem: stanovení diagnózy, prevence, monitorování, léčby nebo mírnění choroby, stanovení diagnózy, monitorování, léčby, mírnění nebo kompenzace poranění nebo zdravotního postižení, vyšetřování, náhrady nebo modifikace anatomické struktury nebo fyziologického procesu, nebo kontroly početí, a který dosahuje funkce nikoli farmakologickým, imunologickým nebo metabolickým účinkem.

32
EU legislativa řídící ZP
Směrnice Rady 93/42/EHS o zdravotnických prostředcích Směrnice Evropského parlamentu a Rady 2007/ 47/ES o aktivních implantabilních zdravotnických prostředcích Směrnice 98/79/ES Evropského parlamentu a Rady o diagnostických zdravotnických prostředků in vitro

33
ČR legislativa řídící ZP
Zákon 123/2000 Sb. o zdravotnických prostředcích, poslední úprava – zákon č. 196/2010 Nařízení vlády ČR č. 336/2004 Sb., kterým se stanoví technické požadavky na zdravotnické prostředky Nařízení vlády č. 453/2004 Sb. v platném znění, kterým se stanoví technické požadavky na diagnostické zdravotnické prostředky in vitro. 154/2004 Sb. Nařízení vlády, kterým se stanoví technické požadavky na aktivní implantabilní zdravotnické prostředky 316/2000 Sb. Vyhláška Ministerstva zdravotnictví, kterou se stanoví náležitosti závěrečné zprávy o klinickém hodnocení zdravotnického prostředku

34
Normy významné pro klinické hodnocení ZP / správnou klinickou praxi
ČSN EN ISO 14155 Klinické zkoušky zdravotnických prostředků pro humánní účely - Správná klinická praxe ČSN EN ISO 13485 Zdravotnické prostředky — Systémy managementu jakosti požadavky pro účely předpisů ČSN EN ISO Zdravotnické prostředky — Aplikace řízení rizika na zdravotnické prostředky GHTF – Reportable events during pre-market clinical investigations MEDDEV 2.7/3 Clinical investigation SAE reporting

35
Zákon 123/2000 Sb. o zdravotnických prostředcích (poslední úprava – zákon č. 196/2010)
Zákon upravuje klinické hodnocení ZP § 8 = Obecná ustanovení § 9 = Etická komise § 10 = Podmínky provádění klinických zkoušek (i) Byla provedena biologicko-bezpečnostní zkouška, potřebná pro ověření určeného účelu použití zdravotnického prostředku, (ii) je prokázána bezpečnostně-technická nezávadnost použití zdravotnického prostředku s přihlédnutím k jeho technickému stavu, předpisům upravujícím bezpečnost a ochranu zdraví při práci a předpisům v oblasti prevence proti vzniku pracovních úrazů, (iii) byly dodrženy etické zásady. § 11 = Dokumentace klinických zkoušek
 Byla provedena biologicko-bezpečnostní zkouška, potřebná pro ověření určeného účelu použití zdravotnického prostředku, (ii) je prokázána bezpečnostně-technická nezávadnost použití zdravotnického prostředku s přihlédnutím k jeho technickému stavu, předpisům upravujícím bezpečnost a ochranu zdraví při práci a předpisům v oblasti prevence proti vzniku pracovních úrazů, (iii) byly dodrženy etické zásady. § 11 = Dokumentace klinických zkoušek.")
36
Nové prvky v zákoně o ZP ve vztahu ke klinickému hodnocení/zkouškám
§ 14 = Způsobilost k provádění klinických zkoušek Zadavatel vybere pro konkrétní klinické zkoušky vhodného poskytovatele zdravotní péče na základě postupu, při kterém posuzuje, zda je systém základního materiálně-technického a personálního zabezpečení požadavků na pracovišti poskytovatele způsobilý k provádění klinické zkoušky konkrétního zdravotnického prostředku. O záměru provést klinické zkoušky musí být před jejich zahájením písemně informován Ústav; v případě multicentrických klinických zkoušek i příslušný úřad členského státu, ve kterém mají být klinické zkoušky prováděny.

37
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 4 Ethical considerations 4.1 General Clinical investigations shall be conducted In accordance with the ethical principles that have their origin in the Declaration Of Helsinki .These principles protect the rights, safety and well-being of human subjects. 4.2 lmproper influence or inducement 4.3 Compensation and additional health care 4.4 Responsibilities 4.5 Communication with the ethics committee (EC)
")
38
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 4 Ethical considerations 4.6 Vulnerable populations 4.7 Informed consent Informovaný souhlas a jeho získání je formulován jako proces naplňující početné požadavky a) - j). lnformed consent shall be obtained in writing from the subject and the process shall be documented before any procedure specific to the clinical investigation. The informed consent form consists of an information form and an informed consent signature form.These two forms can either be combined in one document or separated into two documents.
 - j). lnformed consent shall be obtained in writing from the subject and the process shall be documented before any procedure specific to the clinical investigation. The informed consent form consists of an information form and an informed consent signature form.These two forms can either be combined in one document or separated into two documents.")
39
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 4.7 Informed consent (pokračování) It shall include all aspects of the clinical investigation; avoid any coercion or undue improper influence on, or inducement of the subject to participate; not waive or appear to waive the subjects legal rights; use native non-technical language that is understandable to the subject; provide ample time for the subject to read and understand the informed consent form; include personally dated signatures of the subject and the principal investigator; provide the subject with a copy of the signed and dated informed consent form; ensure important new information is provided to the subject; …
 It shall include all aspects of the clinical investigation; avoid any coercion or undue improper influence on, or inducement of the subject to participate; not waive or appear to waive the subjects legal rights; use native non-technical language that is understandable to the subject; provide ample time for the subject to read and understand the informed consent form; include personally dated signatures of the subject and the principal investigator; provide the subject with a copy of the signed and dated informed consent form; ensure important new information is provided to the subject; …")
40
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 4.7 Informed consent (pokračování) 4.7.3 Special circumstances for informed consent Subject needing legally authorized representatives; Subject unable to read or write Emergency treatments For clinical investigations involving emergency treatments, when prior informed consent of the subject is not possible because of the subject‘s medical condition, the informed consent of the subject‘s legally authorized representative, if present, shall be requested.
 Special circumstances for informed consent. Subject needing legally authorized representatives; Subject unable to read or write Emergency treatments. For clinical investigations involving emergency treatments, when prior informed consent of the subject is not possible because of the subject‘s medical condition, the informed consent of the subject‘s legally authorized representative, if present, shall be requested.")
41
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe Emergency treatments the prospective subject fulfils the emergency conditions and is obviously in a life-threatening situation; no sufficient clinical benefits are anticipated from the currently available treatment; there is a fair possibility that the Iife-threatening risk to the prospective subject can be avoided if the investigational device is used; anticipated risks are outweighed by the potential benefits of applying the investigational device; the Iegally authorized representative cannot be promptly reached and informed.

42
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe Information to be provided to the subject Description and purpose of clinical investigation Potential benefits Risks and inconveniences Alternative procedure(s) Statement confirming that subject participation is confidential as well as records identifying the subject Statement confirming that the subject understands that regulatory authorities and others defined bodies will have direct access to medical records pokračování
 Statement confirming that subject participation is confidential as well as records identifying the subject. Statement confirming that the subject understands that regulatory authorities and others defined bodies will have direct access to medical records. pokračování.")
43
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe Information to be provided to the subject Statement indicating that clinical investigation results may be published without disclosing the subject‘s identity Compensation Anticipated expenses, if any, to be borne by the subject for participating in the clinical investigation Information on the role of sponsor‘s representative in the clinical investigation Contact persons Statement on new findings and amendments to CIP + information of personal physícian Info on trial termination

44
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe Informed consent signature the voluntary agreement or refusal to participate a statement declaring that discontinuation at any time a statement with regard to the possible consequences of withdrawal; an acknowledgement of the information provided and confirmation that all the subjecťs questions were answered a statement confirming that the subject or his/her legally authorized representative agrees to the use of subject´s data a statement on direct access of representatives, regulatory authorities and EC representatives to the subject‘s medical records

45
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 5 Clinical investigation planning 5.2 Risk evaluation 5.3 Justification for the design of the clinical investigation 5.4 Clinical investigation pian (CIP) 5.5 Investigator‘s brochure (lB) 5.6 Case report forms (CRFs) 5.7 Monitoring plan 5.8 lnvestigation site selection 5.9 Agreement(s) 5.10 Labelling 5.11 Data monitoring committee (DMC)
 5.5 Investigator‘s brochure (lB) 5.6 Case report forms (CRFs) 5.7 Monitoring plan. 5.8 lnvestigation site selection. 5.9 Agreement(s) 5.10 Labelling Data monitoring committee (DMC)")
46
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 6 Clinical investigation conduct 6.1 General – všechno podle CIP, žádná změna bez souhlasu CA 6.2 lnvestigation site initiation – zadavatel nebo monitor musí provést návštěvy pracovišť zahrnutých do studie 6.3 Investigation site monitoring – zadavatel musí organizovat monitorovací kontroly pracovišť zahrnutých do studie

47
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 6 Clinical investigation conduct 6.4 Adverse events and device deficiencies Podle Annex 10 Medical Device Directives 93/42/EEC Podle Annex 7 Active Medical Device Directive 90/385/EEC All serious adverse events must be fully recorded and immediately notified to all competent authorities of the Member States in which clinical investigation is being performed Musí se hlásit nejen adverse events, ale také deficiencies (vady identity, kvality atd. ZP)
")
48
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 6.5 Clinical investigation documents and documentation 6.5.1 Amendments 6.5.2 Subject identification log 6.5.3 Source documents 6.6 Additional members of the investigation site team 6.7 Subject privacy and confidentiality of data 6.8 Document and data control

49
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 6.8 Document and data control 6.8.1 Traceability of documents and data All documents and data shall be produced and maintained in a way that assures control and traceability. 6.8.2 Recording of data The data reported on the CRFs shall be derived from source documents and be consistent with these source documents. The CIP shall specity which data can be recorded directly in the CRFs. The CRFs shall be signed and dated by the principal investigator or his/her authorized designee(s). 6.8.3 Electronic clinical data systems
 Electronic clinical data systems.")
50
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 6.9 Investigational device accountability Access to investigational devices shall be controlled and the investigational devices shall be used only in the clinical investigation and according to the CIP. The sponsor shall keep records to document the physical location of all investigational devices from shipment of investigational devices to the investigation sites until return or disposal. The principal investigator or an authorized designee shall keep records documenting the receipt, use, return and disposal of the investigational devices

51
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 6.10 Accounting for subjects 6.11 Auditing Audits of the clinical investigation may be conducted by the sponsor or third parties designated by the sponsor to evaluate compliance with the dp, written procedures, this International Standard and the applicable regulatory requirements. These audits may cover all involved parties, systems and facilities and are independent of, and separate from, routine monitoring or quality control functions.

52
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 7 Suspension, termination and dose-out of the clinical investigation 7.1 Suspension or premature termination of the clinical investigation 7.1.1 Procedure for suspension or premature termination The sponsor may suspend or prematureIy terminate either a clinical investigation in an individual investigation site or the entire clinical investigation for significant and documented reasons.

53
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 7.1.1 Procedure for suspension or premature termination If suspicion of an unacceptable risk to subjects arises during the clinical investigation, or when so instructed by the EC or regulatory authorities, the sponsor shall suspend the clinical investigation while the risk is assessed. The sponsor shall terminate the clinical investigation if an unacceptable risk is confirmed. A principal investigator, EC, or regulatory authority may suspend or prematurely terminate participation in a clinical investigation at the investigation sites for which they are responsible.

54
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 7.1.2 Procedure for resuming the clinical investigation after temporary suspension When the sponsor concludes an analysis of the reason(s) for the suspension, implements the necessary corrective actions, and decides to lift the temporary suspension, the sponsor shall inform the principal investigators, the ECs, and, where appropriate, the regulatory authority of the rationale and provide them with the relevant data supporting this decision.
 for the suspension, implements the necessary corrective actions, and decides to lift the temporary suspension, the sponsor shall inform the principal investigators, the ECs, and, where appropriate, the regulatory authority of the rationale and provide them with the relevant data supporting this decision.")
55
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 7.2 Routine dose-out Routine dose-out activities shall be conducted to ensure that the principal investigator‘s records are complete, all documents needed for the sponsor‘s files are retrieved, remaining clinical investigation materials are disposed of, previously identified issues have been resolved and all parties are notified. Uzavření studie se oznamuje (notifikuje) předepsaným způsobem etické komisi a národním kompetentním autoritám.
 předepsaným způsobem etické komisi a národním kompetentním autoritám.")
56
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 7.3 Clinical investigation report a) The clinical investigation report shall be in written form. b) The clinical investigation report shall include identification of the device(s), a description of the methodology and design of the clinical investigation, any deviations from the CIP, data analysis together with any statistics and a critical appraisal of the aims of the clinical investigation. atd.
 The clinical investigation report shall be in written form. b) The clinical investigation report shall include identification of the device(s), a description of the methodology and design of the clinical investigation, any deviations from the CIP, data analysis together with any statistics and a critical appraisal of the aims of the clinical investigation. atd.")
57
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 7.4 Document retention The sponsor and principal investigator shall maintain the clinical investigation documents as required by the applicable regulatory requirement(s). 8 Responsibilities of the sponsor
. 8 Responsibilities of the sponsor.")
58
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe 9 Responsibilities of the principal investigator 9.1 General 9.2 Qualification of the principal investigator 9.3 Qualification of investigation site 9.4 Communication with the EC 9.5 lnformed consent process 9.6 Compliance with the CIP 9.7 Medical care of subjects 9.8 Safety reporting

59
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe Annex A (normative) CIinicaI investigation plan (CIP) A.1 General - A.18 Bibliography Annex B (normative) Investigator‘s brochure (lB) B.1 General - B.6 Regulatory and other references Annex C (normative) Case report forms (CRFs) C.1 General - C.3 Procedural issues
 CIinicaI investigation plan (CIP) A.1 General - A.18 Bibliography Annex B (normative) Investigator‘s brochure (lB) B.1 General - B.6 Regulatory and other references Annex C (normative) Case report forms (CRFs) C.1 General - C.3 Procedural issues")
60
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe Annex D (informative) Clinical investigation report D.1 General - D.13 Annexes to the report Annex E (informative) Essential clinical investigation documents Table E.1 — Essential clinical investigation documents prior to clinical investigation Table E.2 — Essential clinical investigation documents during clinical investigation Table E.3 — Essential clinical investigation documents after clinical investigation
 Clinical investigation report D.1 General - D.13 Annexes to the report Annex E (informative) Essential clinical investigation documents Table E.1 — Essential clinical investigation documents prior to clinical investigation Table E.2 — Essential clinical investigation documents during clinical investigation Table E.3 — Essential clinical investigation documents after clinical investigation")
61
ČSN EN ISO 14155:2011 Klinické zkoušky zdravotnických prostředků pro humánní účely — Správná klinická praxe

62
Informace o klinických zkouškách v USA i ve světě - http://www
ClinicalTrials.gov is a registry of federally and privately supported clinical trials conducted in the United States and around the world. ClinicalTrials.gov gives you information about a trial's purpose, who may participate, locations, and phone numbers for more details. This information should be used in conjunction with advice from health care professionals. ClinicalTrials.gov currently contains 102,416 trials sponsored by the National Institutes of Health, other federal agencies, and private industry. Studies listed in the database are conducted in all 50 States and in 174 countries.

63
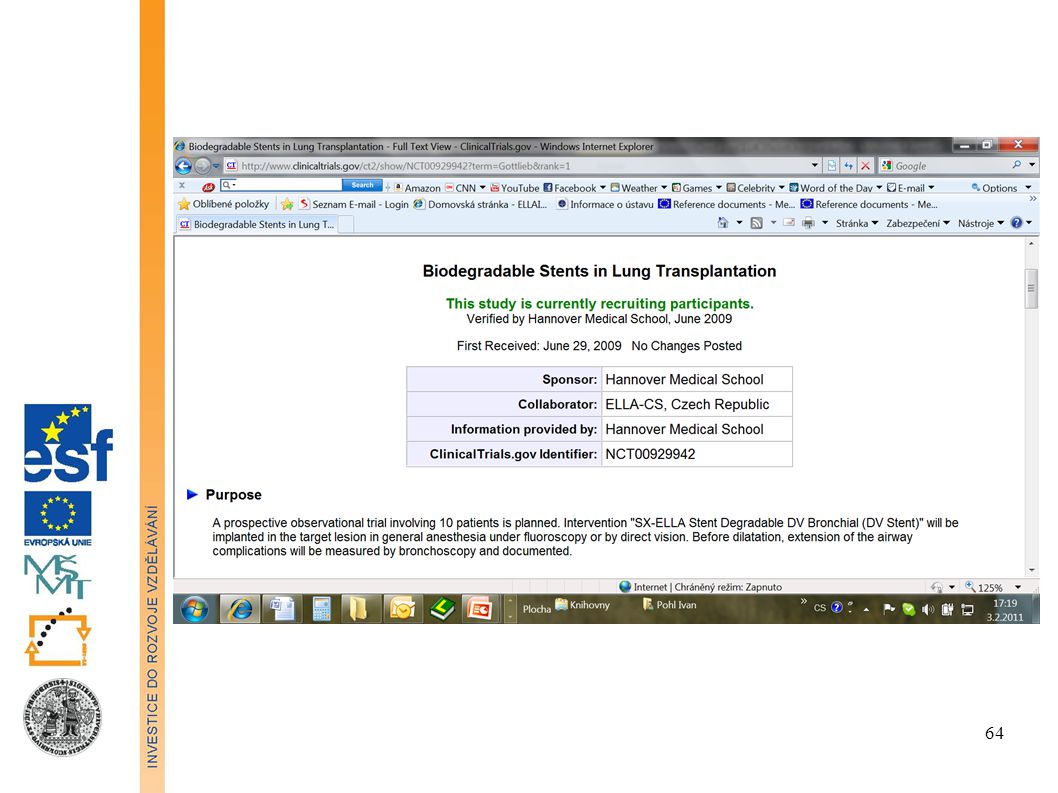
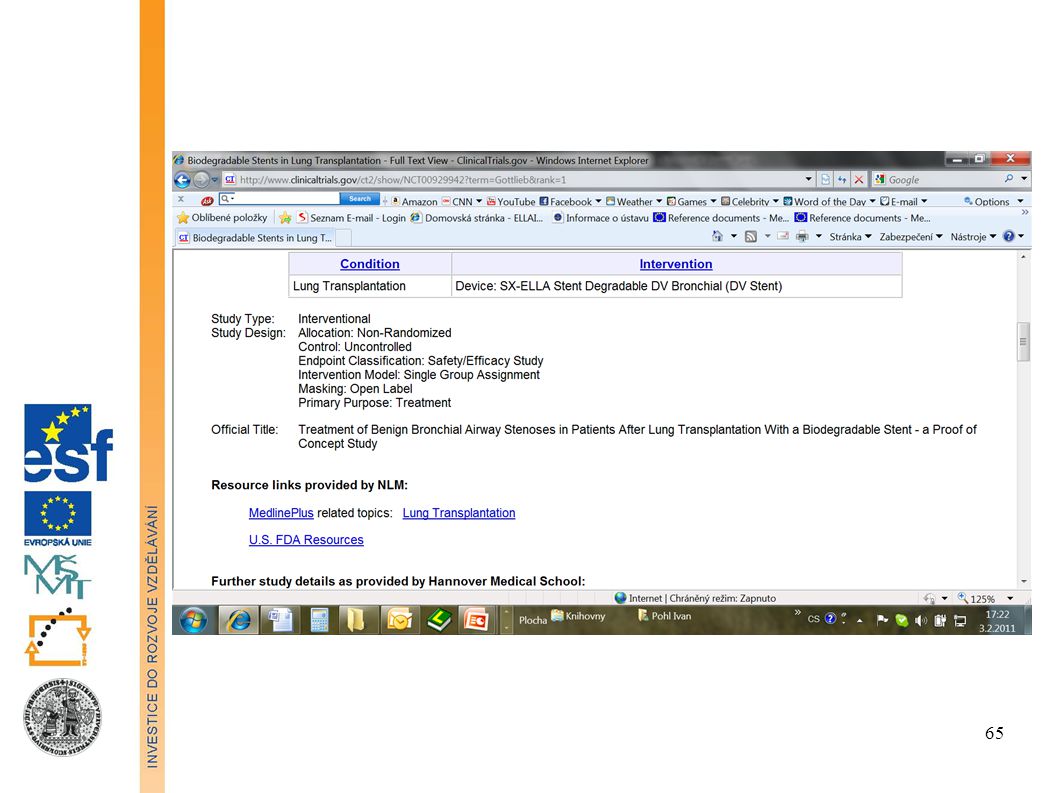
Informace o probíhajících klinických zkouškách
ClinicalTrials.gov is a registry of federally and privately supported clinical trials conducted in the United States and around the world. ClinicalTrials.gov gives you information about a trial's purpose, who may participate, locations, and phone numbers for more details. This information should be used in conjunction with advice from health care professionals. Find trials for a specific medical condition or other criteria in the ClinicalTrials.gov registry. ClinicalTrials.gov currently has 102,416 trials with locations in 174 countries.

68
Děkuji vám za pozornost!

Podobné prezentace